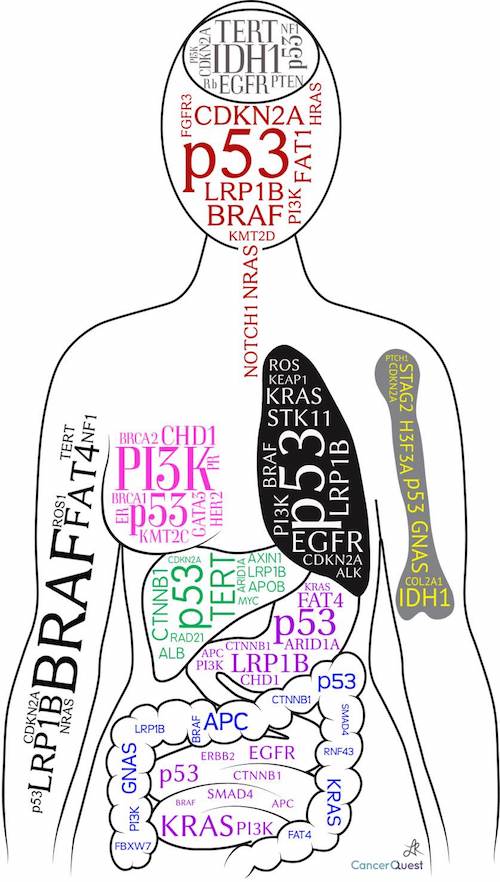
Diagrama mostrando los genes más importantes para algunos cánceres. Mientras más grande se vea el nombre del cáncer, más frecuentemente es defectivo en ese tipo de cáncer.
Introducción
El proceso de la división celular consta de una secuencia de eventos muy controlada. Para que puedan darse con normalidad, estos eventos requieren la transcripción y la traducción de ciertos genes a sus niveles adecuados. Cuando este proceso no ocurre de manera correcta, puede resultar en un crecimiento irregular de la célula. De los aproximados 30 000 genes que actualmente se cree que existen en el genoma humano, existe un pequeño grupo que parece ser particularmente importante en la prevención, el desarrollo y en la progresión del cáncer. Se ha detectado la presencia de variantes defectuosas de los genes pertenecientes a este grupo en muchos tipos de cáncer.
Los genes que han sido identificados hasta la fecha se han categorizado en dos grandes grupos a base de sus funciones normales dentro de la célula.
- Genes cuyos productos protéicos estimulan o aumentan la división y la viabilidad de las células. La primera categoría también incluye a los genes que contribuyen al crecimiento de tumor a través de la inhibición de la muerte celular.
- Genes cuyos productos de proteína previenen la división celular o llevan a la muerte celular directa o indirectamente.
Las versiones normales de los genes en el primer grupo se llaman protooncogenes. Las versiones dañadas o mutadas de estos genes se llaman oncogenes. Note que para distinguir entre estos grupos de genes, los nombres de los protoncogenes se escriben en cursiva o en letras itálicas, mientras que los nombres de sus productos protéicos se escriben en letras regulares. Por ejemplo, TP53 se refiere al gen y P53 a la proteína.
Mientras tanto, los genes en el segundo grupo son de supresión tumoral. Más información sobre los temas discutidos en esta página puede ser encontrada en mayoría de los textos introductorios de biología; nosotros recomendamos Campbell Biology, 11ma edición.2
Temas en esta página:
Oncogenes
Una analogía útil para comprender cómo funcionan los supresores de tumor y los oncogenes es un automóvil. Los protooncogenes representarían el acelerador ya que el coche se debe mover únicamente cuando se lo presiona. En las células normales, tanto las señales internas como la externas controlan la actividad de los oncogenes. En la siguiente animación, estas señales serán representadas por el factor de crecimiento en forma de "X" y por el pie en la porción del video.
Un oncogén defectuoso sería análogo a un acelerador atorado en la posición de "encendido, pues ya no se necesitan señales para activar estos genes. El coche avanzaría independientemente, aun si el acelerador no está siendo presionado.
Esto significa que las células se encuentran en división continua incluso en la ausencia de señales que les indiquen dividirse. Tenemos dos copias de cada gen y para los oncogenes, tan solo una copia defectuosa es suficiente para provocar que una célula se divida sin control.
Los expertos han identificado a varios protooncogenes, y muchos son responsables de proveer señales positivas que llevan a la división celular. Algunos protooncogenes trabajan para regular la muerte celular. Para reiterar, las versiones defectuosas de estos genes, se denominan oncogenes, y pueden provocar que una célula se divida de manera irregular. Este crecimiento puede ocurrir en la ausencia de señales normales que promueven el crecimiento como los factores de crecimiento. Un rasgo clave de la actividad de los oncogenes es que una sola copia alterada puede llevar al crecimiento irregular. A diferencia de los oncogenes, los genes de supresión tumoral deben tener defectos en AMBAS copias genéticas para provocar una división celular anormal.
Los protooncogenes que han sido identificados hasta la fecha tienen muchas funciones en la célula. Independientemente de las diferencias en sus roles normales, todos estos genes contribuyen a una división celular irregular si están presentes en una forma mutante (oncogénica). Las proteínas mutantes a veces retienen algunas de sus capacidades, pero no logran responder a las señales que regulan la variante normal de la proteína.
El cáncer suele depender de un cierto número de mutaciones en oncogenes 'impulsores'. Estas mutaciones son los cambios más importantes que hacen que el cáncer progrese. TODOS los cánceres pasan por muchos cambios adicionales, mutaciones 'pasajeras' que pueden contribuir a cáncer, pero no provienen de los genes principales.
Algunos oncogenes que han sido asociados con varios tipos de cáncer se describen con ás detalle en las siguientes secciones
HER-2/Neu
La proteína HER2 adherida a la porción Fab (fragmento de unión al anticuerpo) del anticuerpo de la Herceptin®.
El HER-2/neu (también llamado erbB-2) es el gen que codifica la producción del receptor del factor de crecimiento epidérmico tipo 2 en humanos. Este receptor se encuentra en cantidades moderadas en algunas células normales y tal como el nombre del gen indica, está involucrado en las respuestas celulares a factores de crecimiento. El siguiente gráfico ilustra cómo la unión del factor bajo las condiciones adecuadas puede estimular la división celular.
La actividad del gen HER-2/neu se amplifica en 30% de los casos de cáncer de mama en humanos. El aumento en el número de copias del gen HER-2lneu lleva a un aumento en la expresión de la proteína HER2 sobre la superficie celular, y se presume que lleva a un aumento en la proliferación celular (mostrada debajo). 3 La investigación también indica que la amplificación de este gen afecta la reacción de los tumores a los tratamientos al igual que a su habilidad para crecer y propagarse. La sobreexpresión de este gen puede provocar que un tumor se vuelva más agresivo, no obstante, también puede elevar el nivel de sensibilidad que posee el cáncer hacia algunos agentes de quimioterapia. 4Más sobre la amplificación de gen.
HER-2/Neu y los tratamientos de cáncer
Se han realizado varios estudios para determinar los efectos que produce la proteína HER-2 en las reacciones corporales a la quimioterapia. Un estudio reciente expuso las células de 140 tumores primarios de seno a varias concentraciones de dos distintas combinaciones de quimioterapia. El estudio mostró que las células que manifestaban altos niveles de actividad en el gen HER2/Neu poseían más sensibilidad a los tratamientos quimioterapéuticos. 1Una revisión de un colectivo de ensayos clínicos logró extender este hallazgo al revelar que se manifestó una sensibilidad elevada hacia las quimioterapias a base de antraciclina, como la doxorrubicina, en pacientes con cánceres de seno con niveles altos de actividad en el gen HER2. La quimioterapia ataca a las células que se replican, y la amplificación del HER-2/neu provoca una replicación más rápida. Una posible conclusión sería que debido a su alto índice de división celular, las células cancerígenas que sobreexpresan el HER2 mueren con más eficacia. Aunque los efectos generales que produce la amplificación del HER2/neu varían, sigue siendo un marcador de pronóstico crítico para el cáncer de seno, y podría guiar el proceso del tratamiento.
Históricamente, la sobreexpresión del HER-2/neu se ha asociado con cantidades elevadas de células anormales (diferenciación celular), una supervivencia corta del paciente y con una escasez de receptores hormonales (enfermedad HR-negativa). 5 Por otro lado, los estudios recientes indican que aproximadamente, la mitad de los casos de cáncer de seno HER2-positivo también son HR-positivos. Esto presenta desafíos para los tratamientos dirigidos. 1 Está claro que este protooncogén es importante en el desarrollo de varias formas de cáncer pero la historia se encuentra lejos de estar completa.
Subtipos intrínsecos del cáncer de mama
Mientras que los niveles de la proteína HER2 se han estudiado a profundidad como biomarcadores del cáncer de mama, los receptores de hormonas (abreviado como HR por sus siglas en inglés) - incluyendo al receptor de estrógeno alfa (abreviado como ERα por sus siglas en inglés) a y/o al receptor de progesterona (abreviado como PR por sus siglas en inglés) - son igual de valiosos para predecir pronósticos y para guiar al tratamiento. Motivados por la meta de encontrar a más factores pronósticos, varios investigadores se enfocan en estudiar los subtipos del cáncer de mama caracterizados por sus padrones distinguidos en la actividad genética (expresión). En el análisis genómico de los tumores del cáncer de mama, se identifican a cuatro sub-poblaciones conocidas como los subtipos intrínsecos del cáncer de mama. Desde su descubrimiento en el año 2000, los subitpos intrínsecos se han confirmado e incluyen lo siguiente:
- Luminal A: expresión elevada del receptor de estrógeno, mutaciones infrecuentes del p53, menos proliferación, mejor pronóstico relativo.
- Luminal B: expresión reducida del receptor de estrógeno, mutaciones frecuentes del p53, más proliferación, pronóstico más agresivo comparado con el Luminal A
- Tipo basal: el subtipo que más carece de receptores de estrógeno y de progesterona, y de la HER2 (a esta enfermedad también se la conoce como cáncer de mama triple negativo); mutaciones frecuentes del p53 y más proliferación.
- HER2-enriquecido(HER2-E): expresión elevada del HER-2/new, mutaciones frecuentes del p53, más proliferación
Los subtipos luminales expresan genes asociados con las células secretoras que recubren los conductos mamarios. El padrón de expresión genética que sigue el subtipo basal se asocia con aquellas células que rodean los conductos mamarios. Interesantemente, no todos los tumores HER-2 positivos corresponden al subtipo HER2-E. Similarmente, no todos los cánceres de seno HER2-E producen un exceso de la proteína HER2. A pesar de la importancia que tiene la clasificación de la enfermedad para fines de pronóstico, se han realizado estudios que procuran encontrar las diferencias entre las reacciones corporales a los tratamientos dirigidos, cuyos resultados no han sido conclusivos. Además, el análisis genómico a menudo se limita a examinar a enfermedades recurrentes o metastáticas. Existen otros exámenes médicos, como la inmunohistoquímica (abreviada como IHC por sus siglas en inglés), que producen resultados más fáciles de emplear en el tratamiento/pronóstico de cánceres de menos avanzados.
Tratamiento de anticuerpo y HER2
Los tratamientos de cáncer han sido diseñados para combatir cánceres que sobreexpresan la proteína HER2. Trastuzumab (Herceptin®), de Genentech,es un anticuerpo monoclonal humanizado que se adhiere a la proteína HER2 y bloquea su actividad, previniendo una proliferación celular excesiva. Este proceso es mostrado en la siguiente animación. El Herceptin® ha sido utilizado recientemente en conjunto con la quimioterapia para cánceres de HER-2/neu amplificado. 5, 3 Más sobre Tratamientos para el cáncer de anticuerpos.
RAS
Los productos del gen Ras están involucrados en las vias de señalamiento de las quinasas que controlan la transcripción de genes, procedimiento que regula el crecimiento y la diferenciación celular. La via de señalamiento empieza cuando la proteína ras se adhiere a una molécula particular (GTP) en la célula. La vía concluye cuando la proteína ras rompe la molécula GTP. Alteraciones en el gen ras pueden cambiar la proteína ras para que ya no sea capaz de proceder como debe. Estos cambios pueden causar que la vía siga en proceso continuo 6, estimulando el crecimiento y la proliferación celular. Por lo tanto, la sobreexpresión de ras y su amplificación puede llevar a una proliferación celular continua, un gran paso en el desarrollo del cáncer. 7 La división celular está regulada por un balance de señales positivas y negativas. Cuando la transcripción de ras aumenta, hay un exceso de la proteína del gen dentro de la célula, y las señales positivas para la división celular comienzan a sobrepasar a las señales negativas.
La transformación del ras de un protooncogén a un oncogén usualmente ocurre cuando se presenta una mutación en el mismo. La función alterada puede afectar la célula de distintas formas, pues el ras está involucrado en muchas vías de señalamiento que controlan la división y la muerte celular. Los fármacos anticáncer están siendo desarrollados para que tengan como blanco las vías dependientes del ras. Todavía queda mucho por descubrir antes de que estos fármacos puedan utilizarse. 8
El Ras mutante ha sido detectado en cánceres de muchos orígenes, incluyendo: páncreas (90%), colon (50%), pulmón (30%), tiroides (50%), vejiga (6%), ovarios (15%), mama, piel, hígado, riñón y algunas leucemias. 6 También es posible que en el futuro, el ras pueda ser utilizado para detectar ciertos cánceres. Históricamente, el cáncer pancreático ha sido difícil de diagnosticar. La identificación de mutaciones del ras en el ADN de las células pancreáticas derramadas en las heces puede facilitar la distinción entre la pancreatitis crónica y el cáncer pancreático. 6
MYC
La proteína myc funciona como un factor de transcripción y controla la expresión de varios genes. Se han encontrado mutaciones en el gen myc en distintos tipos de cáncer, incluyendo en el linfoma de Burkitt, en la leucemia de células B y en el cáncer pulmonar. La familia oncogénica de los genes myc se puede activar con una reorganización de genes o mediante la amplificación. Las reorganizaciones de genes involucran la ruptura y la unión entre cromosomas. Este proceso puede involucrar a grandes cantidades de ADN y por ende, puede llegar a afectar a muchos genes. El movimiento de un gen o grupo de genes hacia una ubicación diferente dentro del mismo cromosoma o hacia otro cromosoma a veces lleva a una expresión de gen y función celular alterada.
La translocación cromosómica es un tipo de reorganización de genes, y una translocación entre los cromosomas 8 y 14 ha demostrado resultar en una sobreexpresión de myc y a última instancia el linfoma de linfocitos B. La siguiente animación muestra cómo se ve una translocación entre dos cromosomas distintos.
La cantidad de proteína myc presente en la célula es importante, pues la actividad de la myc está balanceada por otra proteína que se opone a la actividad de la myc. Por lo tanto, un aumento en cualquiera de estas proteínas puede alterar el balance y afectar la división celular. 9
En el video de arriba, el Dr. Gerard Evan habla sobre el rol de la proteína myc en el cáncer. Vea la entrevista completa con el Dr. Evan.
Un aumento en la actividad myc a veces se asocia la con muerte celular programada, sin embargo este procesos parece ser anulado en la presencia de otro oncogén que la previene apoptosis de myc inducida, como el bcl-2 9
SRC
El Src fue el primer oncogén en descubrirse. Fue identificado como el agente de transformación (causante de cáncer) del virus del sarcoma de Rous (RSV), que infecta a pollos y a otros animales. El RSV es un retrovirus que infecta a células y luego inserta sus propios genes en el ADN celular. Esto resulta rápidamente en el desarrollo del cáncer. Al virus es por ende se lo denomina virus de transformación aguda. Cuando están infectados, los pollos desarrollan grandes tumores en dos semanas. Un grupo de investigadores descubrió que la proteína de un gen particular en el RSV causa que las células crezcan de manera anormal. Un protooncogén correspondiente fue encontrado en el genoma humano. El gen humano, cuando activado como oncogén, funciona de manera similar.
La proteína Src es una tirosina quinasa. Las quinasas son enzimas que transfieren grupos fosfatos a moléculas. El aspecto importante de este proceso es que la extracción/adición de fosfatos cambia a las biomoléculas y es una manera clave de regular las actividades celulares. El proceso de la adición/extracción de fosfatos actúa como un interruptor de encender/apagar para controlar la actividad de las moléculas objetivo. Las proteínas src tienen a varias moléculas de objetivo, resultando en la transmisión de señales al núcleo que ayudan a regular la célula. 9
Se conocen por lo menos nueve genes distintos src. Debido a los distintos procesamientos del mARN producido por estos genes, por lo menos 14 distintas proteínas pueden ser producidas. El C-src normalmente se encuentra en la mayoría de las células a nivel bajo, pero se ha visto sobreexpresado en ciertos tipos de cáncer, incluyendo en el neuroblastoma humano, en los carcinomas pulmonares de células pequeñas, en los cánceres de de colon, de pecho, y en el rabdomiosarcoma. 6
Telomerasa: (símbolo del gen: TERT)
Debido a la naturaleza del proceso de replicación del ADN las partes terminales (telómeros), nuestros cromosomas se achican con cada división celular. El encogimiento de los cromosomas sirve para limitar el número de veces que cualquier célula puede dividirse. Cuando los telómeros se acortan hasta llegar a un largo determinado, la célula es incapaz de replicar su ADN sin perder material genético vital. En este punto las células normales entran al estado de senescencia celular, o detención de crecimiento, después del cual no ocurre ninguna divisón celular.
Las células cancerígenas tienen la habilidad de replicarse sin llegar al estado de senescencia. En muchos cánceres la habilidad de dividirse ilimitadamente se logra con la producción de una enzima llamada telomerasa. La telomerasa mantiene las partes terminales de los cromosomas; es decir, ya no se recortan fragmentos y por ende ya no se achican los cromosomas. Tome en cuenta que la telomerasa es una proteína normal presente en las células durante el desarrollo fetal. Sin embargo, en la mayoría de las células de un adulto humano, la telomerasa no está presente, pues el gen que codifica la producción de la enzima no se está expresando (transcribiendo y traduciendo). En algunas células cancerígenas la replicación ilimitada se da gracias a la reactivación del gen que codifica la producción de la telomerasa.
La animación a seguir ilustra la longitud de dos cromosomas distintos. El de la izquierda carece de la telomerasa activa, mientras que el de la derecha cuenta con la enzima activa.
Se piensa que en las células cancerígenas que viven en ausencia de la telomerasa, el achicamiento de los cromosomas es prevenido por otros mecanismos. El mantenimiento de la longitud del telómero permite la división celular ilimitada. El gen que codifica la producción del para el componente activo de la enzima telomerasa, hTERT, se considera un protooncogén ya que una expresión anormal contribuye a un crecimiento celular irregular.
BCL-2
La familia de proteínas Bcl-2 (provenientes del gen 2 del linfoma de células B) está asociada con membranas y la actividad de membrana. Las proteínas Bcl-2 son parte de un sistema complejo de señalización que controla la apoptosis. La apoptosis (muerte celular) puede ser inducida por una variedad de señales incluyendo el daño irreparable del ADN. Esta forma de suicidio celular previene la replicación de células dañadas. Por otro lado, las Bcl-2 prevenienen la apoptosis, 10 por lo tanto, su sobreexpresión puede prevenir la apoptosis aun en células dañadas. Como consecuencia, se induce una división continua de las células mutadas y eventualmente puede llegarse a desarrollar un cáncer. Adicionalmente, la sobreexpresión de las Bcl-2 puede contribuir a la metástasis en ciertos cánceres. 11
Si los controles de la apoptosis pasan por cambios, los fármacos que inducen la apoptosis, no lo funcionarán con tanta eficacia. Por lo tanto, se están desarrollando fármacos que reducen la cantidad de las Bcl-2, permitiendo que otros fármacos anticáncer funcionen eficazmente incluso a dosis. El nucleótido antisentido Genasense, es uno de estos medicamentos que ha demostrado reducir la producción de las Bcl-2 en la primera fase de pruebas. Actualmente, están en curso las fases II y III de las pruebas, como un tratamiento suplementario para varios cánceres. 12 Sin embargo, la farmacéutica Genta Pharmaceuticals, no logró adquirir los fondos suficientes para seguir con dicha investigación, que subsiguientemente fue obligada a cerrar. Más sobre los fármacos antisentido.
Para agregar, existen fármacos que indirectamente reducen la cantidad de las proteínas Bcl-2, como el ácido retinóico todo trans, el paclitaxel, la vincristina y el docetaxel. Estos fármacos a veces se combinan con otros agentes quimioterapéuticos durante el tratamiento. Nuevos métodos, todavía no probados en humanos, incluyen la adhesión de péptidos Bcl-2 que desactivan a la proteína mientras que la antimicina A que se une a proteínas relacionadas con las Bcl-2. 13
Ya que la sobreexpresión de las Bcl-2 puede afectar al éxito del tratamiento del cáncer, es importante que tanto el médico y como el paciente conozcan la cantidad de Bcl-2s en el cuerpo del paciente. Este protooncogén puede ser activado como oncogén por una translocación que provoca la sobreexpresión del gen, y un aumento en la cantidad de proteínas Bcl-2 se ha visto en muchos tipos de cánceres. 14
EGFR
Tabla de Oncogenes
| Oncogén | Función/Activación | Cáncer* | Referencias |
| ABL |
Promueve el crecimiento celular a través de la actividad de la tirosina quinasa.
|
Leucemia mieloide crónica
|
17, 18 |
| AF4/HRX |
La fusión afecta al factor de transcripción de hrx/ metiltransferasa. El hrx también es conocido como MLL, ALL 1 y HTRX1 |
Leucemias agudas | 19 |
| AKT-2 |
Codifica la producción de la proteína-serina/treonina quinasa |
Cáncer de ovarios | 19, 20 |
| ALK |
Codifica la producción de un receptor de tirosina quinasa. |
Linfomas | 21 |
| ALK/NPM |
La translocación produce una proteína fusionada con nucleofosmina (npm)
|
Linfomas de células grandes | 21 |
| AML1 |
Codifica la producción de un factor de transcripción |
Leucemia mieloide aguda | 19 |
| AML1/MTG8 |
Nueva proteína de fusión creada por una translocación. |
Leucemias agudas | 19 |
| AXL |
Codifica la producción receptor de tirosina quinasa. |
Cánceres hematopoyéticos | 19 |
| BCL-2, 3, 6 |
Bloquea la apoptosis (muerte celular programada) |
Linfomas de células B y leucemias | 18, 19 |
| BCR/ABL |
Nueva proteína creada por la fusión de bcr y abl induce un crecimiento celular irregular. |
Leucemia mieloida crónica y linfática aguda |
17, 18 |
| MYC/c-MYC |
Factor de transcripción que promueve la proliferación celular y la síntesis del ADN |
Leucemia: carcinomas de pecho, de estómago, de pulmón, cervical, y de colon; neuroblastomas y glioblastomas. |
22 |
| MCF2/DBL |
Factor de intercambio del nucleótido Guanina |
Linfoma difuso de células B | 19 |
| DEK/NUP214 |
Nueva proteína creada por fusión |
Leucemia mieloide aguda |
19 |
| E2A/PBX1 |
Nueva proteína creada por fusión |
Leucemia aguda de células pre B |
19 |
| EGFR |
Receptor de superficie celular que provoca el crecimiento celular a través de la actividad de la tirosina quinasa. |
Carcinoma espinocelular | 17, 18 |
| ENL/HRX |
Proteína de fusión creada |
Leucemias agudas | 19 |
| ERG/TLS |
Proteína de fusión creada por la translocación t(16:21). La proteína erg es un factor de transcripción. |
Leucemia mieloide | 18, 19 |
| ERBB |
Receptor de superficie celular que estimula el crecimiento celular a través de la actividad de la tirosina quinasa. |
Glioblastomas y carcinomas de células escamosas |
17 |
| ERBB-2 | Receptor de superficie celular que estimula crecimiento celular a través de la actividad de la tirosina quinasa.; también conocido como HER2 o neu. |
Carcinomas de pecho, glándula salival y de ovarios. |
17, 20 |
| ETS-1 | Factor de transcripción | Linfoma | 23 |
| EWS/FLI-1 | Proteína de fusión creada | Sarcoma de Ewing | 18, 23 |
| FMS | Tirosina quinasa | Sarcoma | 24 |
| FOS | Factor de transcripción para API | Osteosarcoma | 18 |
| FPS | Tirosina quinasa | Sarcoma | 24 |
| GLI | Factor de transcripción | Glioblastoma | 25 |
| GSP | Proteína G asociada a la membrana | Carcinoma de tiroides | 7 |
| HER2/NEU |
Sobreexpresión de una quinasa de señalización causa la una amplificación de gen. |
Carcinomas de pecho y cervical |
17 |
| HOX11 | Factor de transcripción |
Leucemia aguda de células T |
19 |
| HST |
Codifica la producción del factor de crecimiento de firoblasto. |
Carcinomas de pecho y células escamosas. |
19 |
| IL-3 | Molécula de señalización de la célula |
Leucemia aguda de células pre B |
19 |
| INT-2 | Codifica la producción del factor de crecimiento de firoblasto. |
Carcinomas de pecho y células escamosas |
19 |
| JUN | Factor de transcripción para API | Sarcoma | 20 |
| KIT | Tirosina quinasa | Sarcoma | 20 |
| KS3 |
Factor de crecimiento codificado del virus de Herpes |
Sarcoma de Kaposi | 7 |
| K-SAM |
Receptor de factor de crecimiento de firoblasto |
Carcinomas de estómago | 19 |
| LBC | Factor de intercambio del nucleótido Guanina | Leucemias mieloides | 7 |
| LCK | Tirosina quinasa | Linfoma de células T | 7 |
| LMO1, LMO2 | Factores de transcripción | Linfoma de células T | 7 |
| L-MYC | Factor de transcripción | Carcinomas de pulmón | 19 |
| LYL-1 | Factor de transcripción | Leucemia aguda de células T | 19 |
| LYT-10 | Factor de transcripción. También llamado NFκB2 | Linfoma de células B | 7 |
| LYT-10/Cα1 |
Proteína de fusión formada por la translocación (10;14)(q24;q32) de lyt-10 junto al locus de inmunoglobulina C alfa 1. |
19 | |
| MAS | Receptor de la angiotensina | Carcinoma mamario | 7 |
| MDM-2 |
Codifica la producción de una proteína que inhibe y lleva a la degradación de p53 |
Sarcomas | 19 |
| MLL |
Factor de transcripción/metiltransferasa (también llamada hrx y ALL 1) |
Leucemia mieloide aguda | 18 |
| MOS | Serina/treonina quinasa | Cáncer de pulmón | 19 |
| MTG8/AML1 |
Fusión de un represor de transcripción a un factor a un factor de transcripción. AML 1 se conoce como como RUNX1. |
Leucemias agudas | 19 |
| MYB | Factor de transcripción | Carcinoma de colon y leucemias | 19 |
| MYH11/CBFB |
Nueva proteína creada por la fusión entre factores de transcripción vía una inversión en el cromosoma 16. |
Leucemia mieloide aguda | 19 |
| NEU | Tirosina quinasa. También llamada erbB-2 o HER2 |
Glioblastomas y carcinomas de células escamosas |
17 |
| N-MYC |
Proliferación celular y síntesis de ADN |
Neuroblastomas, retinoblastomas, y carcinomas de pulmón | 17 |
| OST | Factor de intercambio del nucleótido Guanina | Osteosarcomas | 7 |
| PAX-5 | Factor de transcripción | Linfoma de células B linfoplasmocitoide | 7 |
| PBX1/E2A |
Proteína de fusión formada vía la translocación de t(1:19). Factor de transcripción |
Leucemia aguda de células pre B | 19 |
| PIM-1 | Serina/treonina quinasa | Linfoma de células T | 7 |
| PRAD-1 |
Codifica la producción de la ciclina D1. Involucrada en la regulación del ciclo celular. |
Carcinomas de pecho y de células escamosas |
19 |
| RAF | Serina/treonina quinasa | Muchos tipos de cáncer | 19 |
| RAR/PML | Proteína de fusión creada por una translocación de t(15:17). Receptor de ácido retinóico | Leucemia promielocítica aguda | 17 |
| RAS-H |
Proteína G. Transducción de señalización. |
Carcinoma de vejiga | 19 |
| RAS-K | Proteína G. Transducción de señalización. |
Carcinoma de pulmón, ovarios y vejiga |
19 |
| RAS-N | Proteína G. Transducción de señalización. | Carcinoma de pecho | 19 |
| REL/NRG | Proteína de fusión creada por supresión en el cromosoma 2. Factor de transcripción. | Linfoma de células B | 19 |
| RET | Receptor de superficie celular. Tirosina quinasa |
Carcinomas de tiroides, Neoplasia endócrina múltiple tipo 2 |
17 |
| RHOM1, RHOM2 | Factores de transcripción | Leucemia aguda de células T | 19 |
| ROS | Tirosina quinasa | Sarcoma | 7 |
| SKI | Factor de transcripción | Carcinomas | 7 |
| SIS | Factor de crecimiento. | Glioma, fibrosarcoma | 7 |
| SET/CAN |
Proteína de fusión creada por una reorganización del cromosoma 9. Localización de proteína. |
Leucemia mieloide aguda | 7 |
| SRC | Tirosina quinasa | Sarcomas | 19 |
| TAL1, TAL2 | Factor de transcripción.TAL1 también es llamado SCL | Leucemia aguda de células T | 19 |
| TAN-1 |
Forma alterada de Notch (un receptor celular) formado por una translocación t(7:9) |
Leucemia aguda de células T | 19 |
| TIAM1 | Factor de intercambio del nucleótido Guanina | Linfoma de células T | 7 |
| TSC2 |
Activador de GTPasa |
Tumores renales y cerebrales | 7 |
| TRK | Receptor de Tirosina quinasa | Carcinomas de colon y de tiroides | 19 |
* Los tipos de cáncer nombrados en esta columna son aquéllos que están predominantemente asociados con cada oncogén pero ésta no es una lista completa.
Para información sobre estos y otros genes por favor visite la página del Proyecto de Anatomía del Genoma del Cáncer.
Resumen de Sección: Oncogenes
Oncogenes
- Los oncogenes son las formas mutadas de los genes celulares normales (proto oncogenes).
- Los productos protéicos de los protooncogenes estimulan la división celular y/o la muerte celular.
- Los protooncogenes se pueden comparar con el acelerador en un coche.
- Normalmente, señales internas y externas regulan estrictamente la actividad de los protooncogenes, sin embargo los oncogenes son defectuosos y están "encendidos" incluso cuando no reciben señales apropiadas.
- Los oncogenes también ayudan a las células a ignorar señales negativas que prevendrían que una célula saludable se divida
- Los oncogenes pueden causar que las células se dividan continuamente incluso en la ausencia de señales promotoras del crecimiento.
- La siguiente lista describe distintos roles celulares de algunos de los muchos oncogenes conocidos:
- HER-2/neu
- HER-2/neu codifica la producción de un receptor de superficie celular que puede estimular la división celular.
- El gen HER-2/neu es amplificado en un 30% de cánceres de mama en humanos.
- RAS
- Los productos del gen Ras están involucrados caminos de señalamiento de quinasa que controlan la transcripción de los genes, regulando el crecimiento y la diferenciación de la célula.
- La sobreexpresión y la amplificación del RAS puede llevar a una proliferación celular continua.
- MYC
- La proteína Myc es un factor de transcripción y controla la expresión de varios genes.
- Se piensa que la Myc está involucrada en la evasión del mecanismo de muerte celular.
- Los oncogenes MYC pueden ser activados por una reorganización o amplificación de genes.
- SRC
- El SRC fue el primer oncogén descubierto.
- La proteína Src es una quinasa tirosina que regula la actividad celular.
- hTERT
- hTERT codifica para una enzima (telomerasa) que mantiene las terminales de los cromosomas.
- En la mayoría de las células normales la telomerasa tan solo está presente durante el desarrollo fetal.
- La activación de hTERT en las células adultas les da la habilidad de dividirse indefinidamente.
- hTERT codifica para una enzima (telomerasa) que mantiene las terminales de los cromosomas.
- BCL-2
- La proteína Bcl-2 trabaja para prevenir la muerte celular (apoptosis).
- La sobreexpresión de BCL-2 permite la división continua de células mutadas.
- HER-2/neu
Conozca el flujo: Oncogenes
"Conozca el flujo" es un juego didáctico para que ponga a prueba sus conocimientos. Para jugar:
- Arrastre las opciones apropiadas de la columna derecha y colóquelas en orden en las cajas de la izquierda. Note que tan solo utilizará cinco o seis opciones para completar el juego.
- Cuando termine, haga click en "Checar" para ver cuántas obtuvo correctas.
- Para respuestas incorrectas, haga click en "Descripción" para revisar la información sobre los procesos.
- Para intentar de nuevo, escoja "Reiniciar" y vuelva a comenzar.
Please visit us on a larger screen to play this game.
Mira las moléculas en el siguiente diagrama para ver las funciones de los oncogenes descritos anteriormente. Muchos otros oncogenes tienen actividades similares a las mostradas.

Genes de supresión tumoral
Los genes de supresión tumoral son clave para varios procesos en la célula, como la regulación de la transcripción, la reparación del ADN y la comunicación de célula a célula. La pérdida de la función de estos genes lleva a un comportamiento celular anormal.
Continuando con la analogía de la sección de los oncogenes, los supresores de tumor pueden ser comparados con el sistema de frenos de un coche. Si compara a ambas copias de cualquier gen de supresión tumoral con los dos frenos presentes en un carro, entonces la analogía es bastante buena. Cuando las dos copias de un gen supresor de tumor están funcionando (representado en la animación por los genes más claros y las señales de alto) la célula puede dejar de dividirse (el coche puede dejar de moverse).
Aunque se presenten defectos en una copia del gen supresor de tumor, aun queda una copia funcional. Sería como frenar a un coche con tan sólo el freno de pie, en vez de usar el freno de mano también. No funciona al 100%, pero sigue funcionando. Por ende, las células con una copia defectuosa de un gen de supresión tumoral aun pueden controlar su división celular. Cuando la segunda copia de la célula se pierde, la célula pierde la habilidad de prevenir la división.
Un muy buen ejemplo de este principio será discutido en la sección del supresor de tumor del retinoblastoma (Rb) en las siguientes secciones.
Todos los cánceres demuestran alteraciones en varios supresores de tumores y/u oncogenes. En las células normales, estos dos grupos de proteínas trabajan en conjunto para regular la división celular mientras que en células cancerígenas, los controles ya no funcionan apropiadamente.
Ya que estos genes son muy importantes para el desarrollo del cáncer, las siguientes secciones van a examinar algunos supresores de tumor en particular y a los cánceres con los cuales están asociados. El número de genes involucrados en estos procesos aumenta casi a diario; los que presentamos aquí son algunos de los mejor estudiados hasta la fecha y dan una buena introducción a algunas de las funciones celulares que se trastornan a causa del cáncer.
Genes de supresión tumoral: p53
El gen p53 (o TP53) fue descubierto en el 1979 y hasta la fecha es uno de los genes más importantes relacionados al cáncer. Este gen, ubicado en el cromosoma 17, produce una proteína que funciona como un factor de transcripción. Los genes controlados por el p53 son aquellos que participan en la división y en la movilidad de la célula. Como otras proteínas de supresión tumoral, la p53 previene el crecimiento irregular de la célula.
La proteína p53 interactúa directamente con el ADN y con otras proteínas que dirigen acciones celulares. Cuando el ADN presenta daños o alguna otra avería, el p53 tiene el poder de inducir la muerte celular o la apoptosis. El papel crucial del p53 consta en regular algunos procesos celulares; esto explica por qué el gen p53 presenta defectos en acerca de la mitad de todos los tumores, sin importar de qué tipo sean o su origen. 9, 26 Las mutaciones que desactivan al gen p53 pueden ser adquiridas de manera esporádica a lo largo de la vida, o pueden ser heredadas.
El descubrimiento del gen p53
En el año 1979, un grupo de científicos descubrió la existencia de nueva proteína. Esta proteína, que lograba unirse a una proteína transformante (antígeno grande T) proveniente del virus Simian 40 (VS40), era más prevalente en aquellas células afectadas (inmortalizadas y posiblemente tumorígenas) por este virus que en células normales. La proteína y su gen correspondiente se denominaron p53, en referencia al peso de la proteína (53 kilodaltons). El gen p53 se encuentra en el cromosoma 17 en la posición p13.27
Aunque la p53 fue la segunda proteína de supresión tumoral en descubrirse (después de la Rb), los científicos no conocían la función que llevaba a cabo dentro de la célula hasta 10 años después de su descubrimiento. 9Ya que la p53 estaba presente a niveles elevados en aquellas células afectadas por el virus, los expertos inicialmente presumieron que actuaba como un oncogén. 28 Este creencia fue apoyada por varias investigaciones tempranas, en las cuales, se halló que cuando el gen p53 se transfirió al interior de las células, ocurrieron algunos cambios y transformaciones. Sin embargo, posteriormente, los expertos descubrieron que el gen p53 que había sido transferido en realidad era un mutante del mismo. Como resultado, se concluyó que una función normal del gen p53 consta en prevenir mutaciones y transformaciones celulares. 28, 9
Varias evidencias apoyan a la conclusión que este gen es un supresor tumoral. Desde el año 1989, las investigaciones acerca del p53 han progresado con estabilidad. Esta proteína se ha visto envuelta en varios procesos celulares. No obstante, aun existen desacuerdos en relación a la confiabilidad del uso del p53 como un instrumento clínico, como en la detección de células cancerígenas por ejemplo.29 Algunas de las investigaciones actuales se enfocan en la posibilidad de reparar o reemplazar al gen cuando se daña.30
Función del p53
La proteína p53 tiene un rol fundamental dentro de la célula, por ende, normalmente se la encuentra en todos los tipos de células. La proteína se ubica en el núcleo donde funciona como un factor de transcripción. La proteína p53 pertenece a una amplia red de proteínas que "monitorean" la salud de la célula y del ADN celular. La proteína p53 es la conductora de un sistema bien coordinado que detecta y controla cualquier daño celular. Cuando se percibe algún daño, la proteína p53 determina si vale la pena reparar el daño, o si se debe iniciar el proceso de la muerte celular (apoptosis). 31
Ya que esta funciona como factor de transcripción, la p53 estimula la transcripción dirigida de un grupo de genes. Entre ellos, el más importante es el p21. El producto del gen p21 es un regulador negativo de las quinasas dependientes de ciclinas, enzimas críticas para el avance del ciclo celular y la división de la célula.9 Cuando se realza la transcripción del gen p21, la p53 impide la proliferación celular. Esta suspensión en la actividad celular, le brinda la oportunidad a las células para llevar a cabo cualquier reparación que sea necesaria. Si ha ocurrido un daño sustancial en el ADN, la proteína p53 puede ayudar a inducir la muerte celular. La muerte de una célula que ha incurrido como consecuencia de un daño importante en el ADN es beneficioso para el organismo, pues evita que las células con mutaciones nocivas se proliferen.
Como se comentó en la introducción de la sección, todas las células cancerígenas tienen mutaciones en los genes de supresión tumoral y en los oncogenes. Cuando la proteína p53, la "guardiana del genoma", está ausente, la célula se encuentra más propensa a la acumulación de daños adicionales en el ADN, y como resultado, la división celular ocurre en varias células que contienen daños en su ADN.
La mutación del gen p53 es uno de los cambios genéticos más frecuentes en las células cancerígenas. Además de las mutaciones que ocurren durante el crecimiento y el desarrollo de individuos (mutaciones esporádicas), existen formas de cáncer asociadas con la herencia de un p53 con defectos. Uno de estos síndromes, el síndrome de Li-Fraumeni, está asociado con una amplia variedad de cánceres. 32 Por otro lado, varios virus han desarrollado mecanismos para desactivar a la proteína p53, como el virus de papiloma humano, el agente causante del cáncer cervical.
Ya que esta proteína es vital para la regulación de la división celular, una gran parte de las investigaciones actuales se enfoca a desarrollar un método seguro para restaurar la función del gen p53.33
En el 2014, un grupo de investigadores descubrió que una versión alterna del p53, en la cual el corte y el empalme se dieron de manera anormal. A esta forma mutada se la denomina p53Ψ y su presencia dentro de la célula INDUCE el desarrollo y la propagación del cáncer. Este estudio se llevó a cabo empleando cultivos celulares en conjunto con anticuerpos provenientes de ratones y conejos.
Las anormalidades en la p53 y el desarrollo del cáncer
Una célula sin una proteína p53 funcional puede o no puede convertirse en cancerígena, pues una célula con una función normal de la p53 aun puede experimentar la formación de un crecimiento cancerígeno. Como se ha mencionado en la sección sobre las mutaciones, para que la célula se transforme a una célula de cáncer, deben ocurrir varios cambios en el ADN. Una de las funciones de p53 consta en controlar al ADN de la célula. En conjunto con proteínas, la p53 ayuda a reconocer daños y a efectuar reparaciones en el ADN dañado. Las reacciones al ADN dañado incluyen a la reparación, la detención de la división celular, y a la muerte celular. Un daño en el gen p53 aumenta la probabilidad de que un individuo desarrolle un cáncer. Recuerde que al ser un gen de supresión tumoral, ambas dos copias del gen p53 deben estar desactivadas para que se estimule un crecimiento cancerígeno. Existen varias maneras por las cuales que el gen p53 se desactiva, descritas a seguir.
Mutaciones
Los cambios que pueden presentarse cambios en el gen p53, pueden ejercer efectos variados en la actividad del gen, dependiendo de la ubicación de la mutación.
-
Las mutaciones pueden ocurrir en las regiones regulatorias de los genes. Estas partes del genoma gobiernan la frecuencia y el momento en el cual se transcribe a un gen (a esta región se la llama el promotor). Una mutación en la región del promotor puede causar en una reducción en la cantidad o la ausencia de la proteína p53 en la célula.34
-
Las mutaciones que ocurren en las regiones que codifican la producción de proteínas pueden alterar la expresión del gen (or la actividad del producto protéico) en varias formas:
- La actividad de la p53 como un factor de transcripción se puede ver reducida. Como resultado, la expresión de los genes regulados por la p53 se vería afectada, como en el gen p21(produce una proteína que está envuelta en la regulación de ciclo celular), el gen Bax (produce una proteína involucrada en la inducción de la apoptosis) y la proteína trombospondina-1 (un inhibidor de angiogenes).35, 36
- Se puede presentar un cambio en el gen p53 que lo hace mas susceptible a la degradación. Si las proteínas p53 en las células se desintegran a una velocidad mayor a la normal, no podrán realizar sus funciones como supresores de tumores.37
Desactivación viral
Una de las funciones de la p53 es en 'guardar' el genoma. La infección con un virus provoca la introducción del ADN viral dentro de las células. La p53, en conjunto con otras proteínas, es responsable por la reacciones inmunes que lleva a cabo la célula al detectar la presencia de un ADN extraño. Las reacciones incluyen la detención de la división celular y la muerte celular. Con fines de interrumpir a estos procesos, varios virus han evolucionado formas de desactivar a la p53. Un ejemplo sería el virus Simian 40 (VS40), cuyas proteínas virales se producen dentro del citoplasma de la célula. A una de estas proteínas se la conoce como el antígeno T grande, cuya función consta en unirse a la proteína p53 para desactivarla. Otros virus como la hepatitis y virus del papiloma humano producen proteínas similares.
La eliminación de un gen p53 funcional de la célula dirige a la célula hacia la división continua, incluso en la presencia de daños en el ADN. En la ausencia del gen p53, la inestabilidad genética se encuentra propensa a crecer a causa del aumento de mutaciones y de la aneuplodía. El incremento del daño genético conduce a la acumulación de defectos en los supresores tumorales y en los oncogenes.
Genes de supresión tumoral: el gen Retinoblastoma (Rb)
El gen retinoblastoma (Rb) codifica la producción de una proteína cuya presencia altera la actividad de los factores de transcripción. A causa de la interacción con los factores de transcripción, la proteína Rb es capaz de indirectamente controlar la expresión genética. Además de esta función, Rb y proteínas relacionadas participan en otras actividades no tan bien estudiadas. A la larga, el Rb y sus parientes contribuyen al control de la división celular. 38
Las mutaciones en el gen Rb son comunes en varios tipos de cáncer. Una de estas enfermedades, y una de las más estudiadas, es el retinoblastoma, un cáncer de ojo por el cual el gen adquirió su nombre. La enfermedad comúnmente aparece en niños pequeños. Se han identificado a dos formas distintas de retinoblastoma.
- La forma esporádica de la enfermedad puede afectar a cualquiera y es dependiente de los cambios genéticos (mutaciones) adquiridos durante la vida del individuo afectado.
- La forma heredada de la enfermedad resulta cuando los individuos afectados heredan una copia defectuosa del gen de uno de sus padres. En estos individuos cada célula contiene una copia normal y una defectuosa del gen. 39
Tal como con otros genes de supresión tumoral, el fenotipo del cáncer no es aparente a menos que ambas copias del gen estén dañadas. Mientras que es poco probable que la copia funcional del gen Rb se mute en cualquier célula, la abundancia de células en nuestros cuerpos (e incluso en un ojo) hacen que la mutación secundaria necesaria ocurrirá con alta probabilidad. Los individuos con la forma heredada de la enfermedad comúnmente sufren de muchos crecimientos cancerígenos, particularmente los osteosarcomas. Otros tipos de cáncer asociados con la mutación Rb incluyen carcinomas de pulmón, mama y de vejiga. 40
El gen Rb y su asociación con el cáncer
El gen Rb se identificó por primera vez al ser asociada con la forma heredada del retinoblastoma. Este tipo de cáncer principalmente afecta a los ojos y es más común en niños. En la forma heredada, los individuos afectados ya poseen una copia mutada del gen Rb en todas sus células, por ende con tan solo una mutación en la otra copia, la célula carece de función de la proteína Rb. Las mutaciones son raras y la posibilidad de que ocurran en un gen singular es pequeña, no obstante el gran número de células en nuestros cuerpos aumenta la probabilidad de que la segunda copia del gen se dañe. Si estas células llegan a crecer descontroladamente, el cáncer puede surgir. En casos de retinoblastomas heredados, el desarrollo de varios tumores es común, pues hay una probabilidad alta de que ocurra una segunda mutación. Por otro lado, en la forma esporádica, uno posee las dos copias funcionales del gen Rb dentro de cada célula; por lo tanto, se requieren dos mutaciones distintas en la misma célula para perder la función de la proteína Rb. Como resultado, aquellos que padecen del retinoblastoma familiar, comúnmente desarrollan un solo tumor. Aquellos que sufren de la forma familiar son más propensos a experimentar recurrencias de tumores.
En pacientes que padecen de osteosarcomas y de un retinoblastoma heredado, se han detectado niveles bajos en la actividad de la proteína Rb. Los osteosarcomas representan casi la mitad de los tumores secundarios identificados en pacientes con la forma heredada de la enfermedad. También se ha demostrado que la función de la proteína Rb afecta la probabilidad de que una mujer desarrolle un cáncer de seno. En circumstancias normales, la proteína Rb regula el punto control G1 del ciclo celular; mientras tanto, varios estudios han demostrado que algunos cánceres de seno carecen de la regulación de este punto de control, provocando que la proteína Rb actúe como un factor contribuyente de la enfermedad. La proteína Rb también se ha visto involucrada en el desarrollo de otros tipos de cáncer, como en el cáncer pulmonar de células pequeñas y en el cáncer pulmonar de células no pequeñas,
Función del Rb
El gen Rb es fundamental para que el ciclo celular funcione con normalidad. Las células responden a una variedad de factores ambientales que las instruyen para crecer, dividirse, descansar o pasar por la apoptosis. Un trastorno en estas señales puede ocasionar un crecimiento irregular en la célula, y a la larga, este se convierte en un cáncer. El control del proceso de la división celular involucra la integración de una variedad de señales.
El producto protéico del gen Rb (pRb) normalmente funciona como un inhibidor de crecimiento al adherirse a los factores de transcripción. Por lo tanto, la proteína Rb indirectamente controla la expresión de una variedad de genes. Algunos de estos genes producen proteínas responsables por llevar a cabo la división celular. Por ello, la actividad del Rb desacelera o frena la división celular. 41 Los cambios en las proteínas reguladoras como la Rb pueden ejercer efectos drásticos en las células, y a la larga sobre el organismo entero. Además de su papel en la regulación del ciclo celular, la Rb también tiene un rol importante en la apoptosis. La apoptosis es una función celular muy importante en la cual una célula dañada se somete a una muerte programada. Si una célula cuenta con mutaciones que no se pueden reparar, la célula se puede eliminar mediante la apoptosis. Este proceso elimina a aquellas células propensas a seguir un crecimiento irregular y cancerígeno. Cualquier alteración en la función celular que reduzca o elimine la activación de la apoptosis puede tener efectos eliminatorios en la población celular. 42 El gen Rb puede ser desactivado a través de distintos tipos de daños genético. Las mutaciones que eliminan por completo la función de la proteína (mutaciones nulas) a veces se ven en células con proteínas Rb no funcionales.
La pérdida de la actividad de la proteína Rb pudiese ayudar a pacientes a que reaccionen saludablemente a la quimioterapia en algunas ocaciones. Múltiples investigaciones han demostrado que aquellos pacientes de cáncer de seno que reciben quimioterapias neo-adyuvantes (quimioterapias administradas antes de una cirugía) reaccionan mejor si carecen de una proteína Rb funcional.43
Más detalles acerca de las funciones del gen Rb
Además de participar en la regulación del crecimiento celular y apoptosis, varios estudios recientes indican que las proteínas relacionadas a la Rb funcionan de distintas maneras dependiendo de la etapa del ciclo celular en la que se presentan y de su ubicación en el núcleo. Para agregar, algunos estudios revelan que las proteínas parecidas a la Rb también regulan la trancripción del rARN y del tARN; esto indica que la proteína Rb logra ejercer el control sobre los eventos trancripcionales y postrancripcionales en las células.44
Aparte de servir como factor de transcripción, se ha observado que varios aspectos de la proteína Rb también contribuyen a la supresión tumoral. La proteína Rb se ha asociado con proteínas que modifican la estructura de la cromatina, como las deacetilasas de histonas por ejemplo. Se presume que estas proteínas alteran la transcripción al eliminar los grupos acetiles en las histonas. Esta modificación resulta en una unión más fuerte entre el ADN y los nucleosomas. 45 Como resultado, se presentan dificultades para los factores de transcripción, como para el E2F, que deben adherirse a sus con sus regiones correspondientes en el ADN. La función de la proteína Rb dentro de este proceso aun no está completamente clara, no obstante múltiples estudios han demostrado que algunas proteínas que modifican las histonas no funcionan apropiadamente en la ausencia de Rb. 46
Genes de supresión tumoral: APC
Las mutaciones en el gen APC (poliposis adenomatosa coli, abreviado por sus siglas en inglés) se encuentran altamente asociadas con los casos esporádicos y heredados del cáncer de colon. La proteína APC, como muchos otros supresores de tumores, controla la expresión de aquellos genes críticos para el proceso de división celular.
Se presume que la mayoría de los casos de cáncer de colon se desarrollan lentamente a lo largo de un periodo de varios años. Se cree que la desactivación del gen APC, ubicado en el cromosoma 5, lleva a un aumento en la proliferación celular y contribuye a la formación de pólipos en el colon. Varias modificaciones genéticas deben ocurrir para que las células de normales del colon se transformen en células capaces de formar tumores. En la mayoría de casos, se cree que una mutación en el gen APC es uno de los primeros pasos de este proceso. Esto se observa indirectamente mediante la examinación de aquellos individuos que han heredado una mutación en una de sus copias del gen APC. Estos pacientes padecen de una enfermedad denominada poliposis adenomatosa familiar, una condición en la cual el colon está lleno de pólipos. Cada pólipo posee la habilidad de desarrollarse en cáncer, por lo tanto aquellos que heredan dicha mutación tienen un riesgo mucho más alto de contraer un cáncer. Si se comparan estas condiciones con aquellas de la forma heredada del retinoblastoma, se puede ver que estas comparten varias similitudes. En vez de necesitar dos mutaciones somáticas dentro de la misma célula para perder la función de APC, estos individuos requieren un solo cambio genético (en cualquier célula) para que se presenten problemas más severos.
Al extraer y examinar células en diferentes etapas del desarrollo del cáncer, se ha establecido el orden más probable que siguen las mutaciones genéticas causantes del crecimiento de varios cánceres de colon. Según este modelo, el gen APC experimenta mutaciones dentro de los primeros pasos, produciendo células altamente propensas a proliferarse. Como resultado, estas células proceden a formar pólipos que pueden desarrollarse en cánceres. 9
Función del gen APC
Ya que la ausencia de la proteína APC provoca un aumento en la división celular, sería lógico que la proteína en su estado natural inhiba la división celular. En efecto, este es el caso. La proteína APC se adhiere a la catenina-beta, un factor de transcripción, formando un complejo; como resultado, la catenina-beta se desintegra. En la ausencia de la proteína APC, se acumula un exceso de catenina-beta en el núcleo. La catenina-beta se adhiere a otra proteína dentro del núcleo para formar un complejo que se une al ADN, activando la transcripción de varios genes, como el oncogén. 47 c-myc. El c-myc por sí solo es un factor de transcripción que promueve la expresión de varios genes que controlan el crecimiento y la división celular. Por lo tanto, una mutación en el gen APC desencadena una serie de eventos que eventualmente provocan un aumento en la división celular. A seguir se encuentra un diagrama que ilustra el rol del gen APC:
Aunque existen muchos otros factores que afectan a la expresión de los genes y a su productos, las mutaciones que ocurren dentro del gen APC parecen estar relacionadas con un aumento en la cantidad de la catenina-beta y del producto proteíco de la c-myc, acelerando la proliferación celular. 48
Varias investigaciones han demostrado que la incorporación de la proteína APC en su estado natural dentro de células de cáncer de colon que carecen de la forma funcional de dicha proteína, ocasiona un retraso en el crecimiento celular del tumor. Estos estudios han revelado que esta disminución en el crecimiento cancerígeno se debe a un aumento en la apoptosis, indicando que la proteína APC regula los controles de la muerte celular y del crecimiento. 49 Por lo tanto, una pérdida de genes funcionales altera el balance entre el crecimiento celular y la muerte celular, que controla la cantidad de células en el cuerpo.
Genes de supresión tumoral: BRCA
Las proteínas BRCA tienen múltiples funciones. Por ejemplo, además de llevar a cabo la reparación de daños en el ADN, también participan en la regulación de la expresión de genes. El gen BRCA-1 está asociado con la activación del gen p53, y por ende del gen p21. Las proteínas BRCA también interactúan con algunos factores de transcripción y otros componentes transcripcionales para controlar la actividad en algunos genes. 50 Cuando los genes BRCA presentan defectos, la reparación del ADN y la regulación celular se encuentran comprometidas. Como resultado del aumento de daños en el ADN, se producen células que acumulan mutaciones en genes clave, llevando a la formación de células cancerígenas. Las células que carecen de genes BRCA en sus estados normales suelen experimentar rupturas cromosómicas, aneuploidías severas y niveles excesivos de centrosomas. Todos estos defectos interfieren con la función y la división normal de la célula.
Al nivel molecular, los genes BRCA-1 y BRCA-2 se encuentran vulnerables a pasar por mutaciones, pues sus estructuras facilitan dicho proceso. Contienen una proporción muy alta de secuencias repetitivas en el ADN,una ocurrencia rara en los genes humanos. El ADN repetitivo puede crear una inestabilidad genómica y ocasionar reorganizaciones. 50
Varias investigaciones han demostrado que la pérdida de productos proteícos provenientes del gen BRCA está asociada con el desarrollo del cáncer heredado y esporádico. 50
El cáncer de ovario y los genes BRCA
Aunque los genes BRCA se nombraron por su participación en el cáncer de seno, las mutaciones en estos genes también se asocian con el cáncer de ovario. Las formas heredadas y esporádicas del cáncer de ovario comparten similitudes, no obstante existen algunas diferencias. El cáncer de ovario heredado tiende a tener una histología serosa, a diferenciarse de manera moderada o pobre, a ser invasivo, y usualmente se lo detecta en sus etapas avanzadas. Por otro lado, aquellos individuos que presentan mutaciones en sus genes BRCA, experimentan una frecuencia más alta de lesiones en las trompas de falopio. Independientemente de la presencia o ausencia de la mutación en el paciente, los tumores benignos o malignos de potencial bajo, no se consideran como precursores del carcinoma de ovario invasivo.51
Función de BRCA
Las mutaciones en los genes BRCA-1 y BRCA-2 están asociadas con algunos cánceres de mama y de ovarios. Estos dos genes tienen funciones distintas dentro de las células. Al igual que otros genes de supresión tumoral, las mutaciones pueden ocurrir espontáneamente o se las puede heredar. Los individuos que heredan una mutación en el BRCA-1 o en el BRCA-2 son más propensos a desarrollar un cáncer de mama. Además, aquellos que presentan mutaciones en los genes BRCA tienen un riesgo de por vida (si llegan a los 85 años de edad) de 80% de desarrollar un cáncer de mama. Por otra parte, el riesgo de desarrollar un cáncer de ovario es de 10-20% cuando las mutaciones ocurren en el gen BRCA-2 y de 40-60% cuando se presentan en el gen BRCA-1. La presencia de estas mutaciones también puede aumentar el riesgo de contraer un cáncer de próstata, pancreático, de colon y otros. El riesgo total para cualquier persona depende de los factores genéticos y ambientales de riesgo a los cuales se expone. Se presume que las mutaciones en los genes BRCA-1 y BRCA-2 están asociadas con un 5-10% de los cánceres de mama.
Los genes BRCA y el estrógeno
Las mutaciones en los genes BRCA han sido asociadas con cánceres en ciertos tejidos, como aquellos en el pecho y en los ovarios. Por lo tanto, es probable que el estrógeno tenga un rol en el desarrollo del cáncer en estos tejidos. Las fluctuaciones en la cantidad del estrógeno, tales como las que se ven en la pubertad, la menstruación, el embarazo y en la menopausia, también se asocian con el desarrollo del cáncer. Una cantidad elevada de estrógeno, particularmente durante la pubertad y el embarazo, aumenta la proliferación de las células epiteliales del seno, lo cual por su parte coloca demandas mayores en las capacidades de reparación del ADN en las células. Cuando aquellas células que no logran reparar daños en el ADN con eficacia se reproducen, la formación del cáncer. En la siguiente animación, el estrógeno (que aparece en rosa) estimula la división celular, produciendo una célula cancerígena. 50
Si un gen BRCA ya se encuentra mutado, una segunda mutación que eliminaría la única copia funcional del gen provoca deficiencias la reparación del ADN dañado. Cuando ambas copias del gen reparador presentan defectos, se aumenta la probabilidad de que la célula adquiera mutaciones que lleven al desarrollo de tumor. En un individuo que ha heredado una copia defectuosa del gen BRCA, TODAS sus células cuentan con el defecto. Por lo tanto, una mutación singular en la segunda copia del gen dentro de cualquier célula puede ocasionar dificultades en la reparación de ADN. Por otro lado, se requieren dos mutaciones separadas para que se desarrolle el cáncer en aquellos individuos que no han heredado un alelo deficiente del gen BRCA. Ambas mutaciones esporádicas deben ocurrir en la misma célula. La ocurrencia de dos mutaciones en la misma célula es rara, por lo cual estos cánceres tienden a presentarse en etapas más avanzadas de la vida.50
Puede encontrar más información acerca de este tema en los capítulos 3, 4, 7 y 9 de La biología del cáncer por Robert A. Weinberg.
Vista Cercana a los Efectos de BRCA en la Supervivencia
Se han realizado varios estudios diseñados con fines de determinar las diferencias en la supervivencia entre los portadores de mutaciones del BRCA con el cáncer y aquellos con desarrollaron el cáncer esporádicamente. Los resultados son un poco contradictorios, probablemente a causa de la variedad de factores involucrados en el diseño de estos estudios, como en el grado de igualdad entre las mutaciones esporádicas y heredadas. Sin embargo, aunque los portadores de la mutación en el gen BRCA suelen tener peores pronósticos (dependientes de las características de su cáncer), tienden a experimentar tazas más altas o iguales de supervivencia comparado con los pacientes con un cáncer esporádico. Se presume que esto se debe a la sensibilidad de los tumores a la quimioterapia, cuyo aumento es causado, en parte, por velocidades elevadas de proliferación celular. Los tumores también son más sensibles a los tratamientos de cáncer como la radiación gamma, el ciplatino y la mitomicina C ya que estos tratamientos causan daños en el ADN que normalmente sería reparado por los productos del gen BRCA funcional. Si los genes BRCA se desactivan, la célula no logra reparar el daño en su ADN eficazmente, ocasionando la muerte celular. Las células sin cáncer en un portador de la mutación de BRCA retiene un gen funcional de BRCA y entonces se encarga de reparar al ADN. 51
Tabla de genes de supresión tumoral:
| Supresor de tumor | Función | Cáncer* | Referencias |
| APC |
Controla la función de factores específicos de transcripción los cuales están involucrados en la tumorigénesis, y el desarrollo y la homeostasis de algunos tipos de célula incluyendo células epiteliales y linfoides. APC también ha sido implicado en la proliferación celular y otras actividades celulares como migración y adhesión. |
Adenomatosa familiar y carcinomas colorrectales no hereditarios.
|
52, 53 |
| BRCA1, BRCA2 |
Reparación de daño en el ADN
|
Cánceres de mama hereditarios; cánceres de ovario. |
54 |
| CDKN2A |
Locus de gen que codifica los supresores de tumor p16 y p14ARF. |
Tumores cerebrales | 52, 55 |
| DCC |
Receptor de netrina 1. Regulación de la proliferación celular y apoptosis del epitelio intestinal. |
Carcinomas colorrectales | 56, 57, 58 |
| DPC4 (SMAD4) |
Factor transcripcional involucrado en el desarrollo; implicado en la metastasis y en la invasión de tumor. |
Tumores colorrectales, neoplasia pancreática
|
59, 60 |
| MADR2/JV18 (SMAD2) |
Modera las señales de los receptores de factores de crecimiento. Asiste en el transporte del SMAD4 hacia el núcleo. |
Cáncer colorrectal | 61, 62 |
| MEN1 |
Codifica la proteína menina que interactúa con factores de transcripción, proteínas de reparación de ADN, proteínas citoesqueléticas y otros. Su función no está definida claramente. |
Neoplasia endócrina múltiple tipo 1
|
63 |
| MTS1 |
Inhibidor de quinasas dependientes de ciclina; regula el pasaje del ciclo celular de G1 a S. |
Melanomas | 64 |
| NF1 |
Proteína de activación de la RAS GTPasa (RAS-GAP) |
Neurofibromatosis tipo 1 | 65 |
| NF2 |
Proteína ERM; organiza la membrana plasmática ensamblando complejos de proteína y vinculándolos a la actina. |
Neurofibromatosis tipo 2 | 66 |
| p53 |
Codifica un factor de transcripción para p21, una proteína que detiene el ciclo celular en la fase G1. p53 integra señales relacionadas con el tamaño celular, la integridad del ADN y la replicación cromosómica. |
Carcinomas de vejiga, seno, colorrectal, esofágico, de hígado, pulmonar, de próstata, y de ovarios; tumores cerebrales, sarcomas, linfomas y leucemias. |
67 |
| PTEN |
Fosfatasa lipídica. Regula la supervivencia celular. |
Síndrome de Cowden; aumento en el riesgo de cáncer de mama y de tiroides. |
53, 68 |
| Rb |
Se une , e inhibe, al factor de transcripción E2F. Detiene la progresión del ciclo celular. |
Retinoblastoma, sarcomas; carcinomas de vejiga, seno, esofágico, de próstata y pulmonar. |
69 |
| VHL |
Regulación del ciclo celular. Puede aumentar la estabilidad y actividad de p53. |
Carcinoma de células renales | 52, 70 |
| WRN |
ADN helicasa y exonucleasa. Involucrada en la reparación de las rupturas de ADN. |
Síndrome de Werner | 53, 71 |
| WT1 |
Factor de transcripción. Rol esencial en el desarrollo. |
Tumores de Wilms (cáncer pediátrico de riñón) |
52 |
* Los tipos de cáncer listados en esta tabla son aquellos que están predominantemente asociados con cada gen de supresión tumoral, mas no es una lista exhaustiva.
Para más información sobre estos genes y otros por favor visite el Proyecto de anatomía del genoma del cáncer.
1. Cooper G. Oncogenes. Jones and Bartlett Publishers, 1995.
2. Vogelstein B, Kinzler KW. The Genetic Basis of Human Cancer. McGraw-Hill: 1998.
Resumen de sección: Genes de supresión tumoral
Supresores de Tumor
- Los productos protéicos de los genes de supresión tumoral pueden prevenir la división celular o provocar la muerte celular directa o indirectamente
- Los supresores de tumor pueden compararse con los frenos de un coche.
- La pérdida de funciones de los supresores de tumor puede llevar a un comportamiento anormal de la célula.
- A continuación se describe la función de algunos genes supresores clave de tumor:
-
- p53
- Un factor de transcripción que regula a los genes que controlan la división celular y la muerte celular.
- Importante en la respuesta celular a daño de ADN.
- Determina si vale la pena reparar los daños que se presentan en el ADN o si se debe proseguir con la muerte celular.
- Rb
- Funciona a través de la alteración de la actividad de los factores de transcripción.
- Contribuye al control de la división celular al ser un inhibidor.
- APC
- La proteína APC se adhiere y estimula la degradación de un factor de transcripción.
- La ausencia de la proteína APC funcional lleva a un aumento en la división celular.
- BRCA
- Las proteínas BRCA tienen funciones múltiples, incluyendo la reparación de daños en el ADN y la regulación de la expresión de genes.
- Las BRCA deficientes perjudican a la reparación del ADN y a la regulación genética
- p53
MicroARN
Los genes son cadenas largas de ADN que codifican información genética en la forma de ARN. Por muchos años, los genes de mayor interés fueron los que se codifican mediante el ARN mensajero (ARNm), sustancia que se utiliza como guía para la producción de proteínas. (Revise nuestra sección acerca de la función de los genes para un resumen general.) Las otras especies de ARNs (ARNt, ARNpno, ARNr) son tan útiles como el mismo ARNm y participan en la producción de proteínas.
En el año 1993 nuevo tipo de ARN fue descubierto dentro de un tipo de gusano; esta nueva molécula era bastante pequeña a pesar de sus altos niveles de actividad 72. Este ARN regulaba la actividad de un grupo distinto de genes. Mientras que las moléculas del ARNm pueden tener miles de nucleótidos de longitud, este ARN de novedad solamente contaba con algunas docenas de nucleótidos de largo. Dentro de una década, muchos otros ejemplos de estos ARNs pequeños fueron descubiertos. Hoy en día, a estas moléculas se las conoce como MicroARN (abreviado como miARN), y se distinguen por su capacidad de controlar diferentes genes en los procesos celulares.
Otra sorpresa surgió cuando los investigadores procuraban descubrir nuevos tipos de miARNs. La información genética de algunos de ellos proviene de su propio gen, pero muchos la contienen dentro de otros genes, generalmente en partes que no se utilizan para crear proteínas (las llamadas regiones no codificantes). También, los miARNs no se producen en una forma funcional, pues requieren varios pasos de procesamiento para finalmente trabajar en conjunto con proteínas y así cumplir con su actividad de regulación de genes.
El siguiente diagrama ilustra dos vías distintas que llevan a la producción de miARNs funcionales. (Gráfico de Wikimedia Commons).
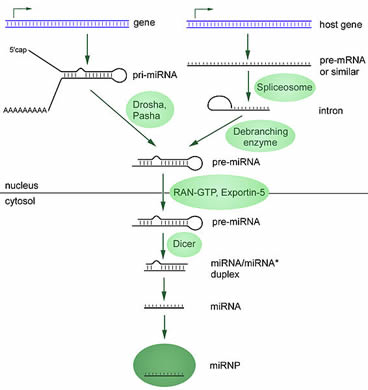
El producto final del proceso de 'maduración' es un ARN corto, combinado con un grupo de proteínas (un miRNP). La función del miRNP consiste en aumentar o disminuir la actividad de algunos genes. Un miRNP maduro puede unirse a un ARNm para prevenir su uso en la producción de proteínas. Estos complejos también pueden causar la destrucción directa de las moléculas a las que se adhieren. Dado que la actividad de nuestros genes está estrictamente controlada para mantener el balance en nuestras células, la asociación entre los defectos que se manifiestan en la producción o en la actividad de los miARNs con varias enfermedades del cuerpo humano, incluyendo el cáncer, no es inesperada. 73, 74, 75, 76
El microARN y el cáncer
Los microARNs (miARNs) se distinguen por su abundancia dentro del cuerpo y al regular a genes involucrados en un amplio rango de actividades celulares. Por ende, cuando ocurren cambios estimulantes o inhibidores en los miARNs, la actividad de los genes que regulan se altera, llevando a cambios visibles como la enfermedad. Por ejemplo, el cáncer es el resultado de la suma de varias modificaciones genéticas que alteran la actividad de los genes, por lo cual es razonable que los cambios en los miARNs lleguen a ejercer sus efectos en el desarrollo y/o propagación del cáncer. De hecho, la investigación acerca de los miARNs es extremadamente activa ya que impacta a muchas áreas diferentes de la biología del cáncer, como la detección, el diagnóstico y el tratamiento.77, 78, 79, 80 Algunas de las áreas del estudio del cáncer afectadas por los miARNs se describen a continuación.
Los miARNs y la prevención del cáncer
Nuestra comida contiene una variedad de compuestos químicos que influye en la actividad de muchos genes, incluyendo a aquellos que codifican la información genética de los miARNs. Por lo tanto, muchas investigaciones se enfocan en los efectos que tienen nuestras dietas en el aumento o en la disminución del riesgo de desarrollar un cáncer.
Los miARNs como oncogenes y supresores de tumores
Ya que los miARNs controlan la actividad de los genes, podrían considerarse como oncogenes o supresores de tumores, dependiendo de su efecto en el crecimiento celular. Los miARNs que normalmente disminuyen la velocidad de la división celular o que causan la muerte celular se clasificarían como supresores de tumores (su ausencia llevaría a un aumento en la división/supervivencia de la célula) y aquellos que normalmente incrementan la división celular o la supervivencia celular serían considerados oncogenes. Hasta la fecha, se han observado varios casos en los cuales los miARNs llevan a cabo dichas funciones en una variedad de distintos tipos de cáncer. 75, 81, 76, 82
Los miARNs como conductores del metabolismo celular tumoral
Ya por varios años se ha conocido que las células cancerígenas dependen de la glucólisis, una ruta metabólica perteneciente a la producción de energía celular, a niveles desproporcionados. A este suceso se lo denomina como el 'efecto Warburg'. Como resultado, las células cancerígenas adquieren más azúcar en comparación con otras células, mientras que los niveles de azúcar en el cuerpo también las afecta irregularmente. Adicionalmente, las células cancerígenas producen más ácido láctico, producto de la glucólisis que puede alterar el ambiente que rodea de las células. En conjunto, estos cambios pueden conducir a la progresión de la enfermedad. Se presume que la actividad de los miARNs provoca el efecto Warburg mediante su influencia en la actividad de supresores tumorales como p53, y oncogenes, incluyendo HIF1A.83, 84, 85
Los miARNs como biomarcadores para la detección y el diagnóstico de cáncer
Un biomarcador es algo que indica la presencia de una enfermedad (o el potencial de la misma) de manera indirecta. Un ejemplo de un biomarcador es el colesterol en la sangre como un indicador de la salud cardiovascular. Los exámenes de sangre como la prueba PSA también son pruebas de biomarcadores. Ya que los miARNs han sido asociados con el cáncer, varias investigaciones procuran determinar si la presencia de los miARNs en la sangre puede servir como un biomarcador del cáncer y por lo tanto podría ser una base para la prueba.86, 78, 79, 80 También se ha propuesto su función como marcadores de la resistencia a fármacos y su uso en el seguimiento del tratamiento.87
Los miARNs como objetivos para el tratamiento de cáncer.
Dado que los miARN regulan la actividad celular, estas moléculas podrían servir como enfoques en el desarrollo de tratamientos de cáncer. Un solo miARN logra controlar a una cantidad extensa de genes, por ende, los fármacos señalados al miARN podrían ser bastante efectivos, pues varias vías metabólicas podrían activarse o desactivarse en su totalidad mediante estos medicamentos.88, 78, 79, 80, 81
Por ejemplo, muchos tipos de cáncer de seno dependen de las hormonas sexuales femeninas como el estrógeno y la progesterona para su crecimiento y supervivencia. Esta observación es la base para el uso de tratamientos anti hormonales como el tamoxifen, el raloxifene y los inhibidores de la aromatasa. En un estudio realizado en el 2012, se halló que la progesterona provocaba que las células cancerígenas se parezcan más a las células madre, dificultando el tratamiento. Se concluyó que este cambio en el comportamiento celular se debía a la supresión de un grupo de miARNs conocido como la familia 29 de miARNs. Hoy en día, muchos estudios buscan encontrar la manera de incrementar la actividad de estos miARNs en las células cancerígenas con la esperanza de revertir los rasgos de células madre.89
Los miARNs como impulsores de la resistencia a medicamentos
A veces, los medicamentos de cáncer logran ser eficientes solamente en las etapas iniciales del tratamiento. Con el tiempo, el paciente puede perder la sensibilidad al medicamento. Este fenómeno de resistencia a los medicamentos es lo que dificulta el tratamiento del cáncer. Por lo tanto, bastantes investigaciones se enfocan en cómo y por qué esta resistencia se desarrolla. Las evidencias recientes indican que los miRNAs ayudan a impulsar este cambio. Cantidades anormalmente excesivas de miRNAs se asocian con la fármacorresistencia. Sin embargo, cuando este exceso se revierte a una cantidad normal, la sensibilidad a los medicamentos puede volver.90
Aprenda más acerca de células madre y el cáncer.
- 1abc
- 2 Urry, L. A., Cain, M. L., Wasserman, S. A., Minorsky, P. V., & Reece, J. B. (2017). Campbell Biology (11th ed.). Pearson.
- 3ab Tsuda H, Akiyama F, Terasaki H, Hasegawa T, Kurosumi M, Shimadzu M, Yamamori S, Sakamoto G. "Detection of Her-2/neu (c-erb B-2) DNA Amplification in Primary Breast Carcinoma." Cancer (2001). 92(12): 2965-2974. [PUBMED]
- 4 Konecny G, Fritz M, Untch M, Lebeau A, Felber M, Lude S, Beryt M, Hepp H, Slamon D, Pegram M. "Her-2/neu Overexpression and in vitro Chemosensitivity to CMF and FEC in Primary Breast Cancer." Breast Cancer Research and Treatment (2001). 69: 53-63. [PUBMED]
- 5ab Spizzo G, Obrist P, Ensinger C, Theurl I, Dunser M, Ramoni A, Gunsilius E, Eibl G, Mikuz G, Gastl G. "Prognostic Significance of Ep-CAM and Her-2/neu Overexpression in Invasive Breast Cancer." Int. J. Cancer (2002). 98: 883-888. [PUBMED]
- 6abcd Ruddon RW. Cancer Biology. Oxford University Press: New York, 1995.
- 7abcdefghijklmnopq Vogelstein B, Kinzler KW. The Genetic Basis of Human Cancer. McGraw-Hill: 1998.
- 8 Ahmadian MR. "Prospects for Anti-ras Drugs." British Journal of Haematology (2002). 116(3-I): 511-518. [PUBMED]
- 9abcdefgh Cooper G. Oncogenes. Jones and Bartlett Publishers, 1995. 151-152, 175-176.
- 10 Strasser A, Huang DC, Vaux DL. "The role of the bcl-2/ced-9 gene family in cancer and general implications of defects in cell death control for tumourigenesis and resistance to chemotherapy." Biochim Biophys Acta. 1997 Oct 24;1333(2):F151-78. [PUBMED]
- 11 Fernandez Y, Gu B, Martinez A, Torregrosa A, Sierra A. "Inhibition of Apoptosis in Human Breast Cancer Cells: Role in Tumor Progression to the Metastatic State." Int. J. Cancer (2002). 101: 317-326. [PUBMED]
- 12 Genta Pharmaceuticals [http://www.genta.com]
- 13 Obasaju C, Hudes GR. "Paclitaxel and docetaxel in prostate cancer." Hematol Oncol Clin North Am. 2001 Jun;15(3):525-45. [PUBMED]
- 14 Gross A. "BCL-2 proteins: regulators of the mitochondrial apoptotic program." IUBMB Life. 2001 Sep-Nov;52(3-5):231-6. [PUBMED]
- 15abc Santos, Gilda Da Cunha, Frances A. Shepherd, and Ming Sound Tsao. "EGFR Mutations and Lung Cancer." Annual Review of Pathology: Mechanisms of Disease 6 (2011): 49-69. [http://www.ncbi.nlm.nih.gov/pubmed/20887192] [PUBMED]
- 16 Seshacharyulu, Parthasarathy et al. ¿Targeting the EGFR Signaling Pathway in Cancer Therapy.¿ Expert Opinion on Therapeutic Targets 16.1 (2012): 15¿31. PMC. [http://www.ncbi.nlm.nih.gov/pubmed/22239438] [PUBMED]
- 17abcdefghij Gustave Roussy, Cytogenomics of cancers: From chromosome to sequence, Molecular Oncology, Volume 4, Issue 4, August 2010, Pages 309-322 [PUBMED]
- 18abcdefgh Carlo M. Croce, M.D."Oncogenes and Cancer". N Engl J Med 2008; 358:502-511. [http://www.nejm.org/doi/full/10.1056/NEJMra072367]
- 19abcdefghijklmnopqrstuvwxyz Cooper G. Oncogenes. Jones and Bartlett Publishers, 1995.
- 20abcd Amplified and Over-expressed Genes in Cancer Table. The Institute of Cancer Research. 2009-2010. [http://www.amplicon.icr.ac.uk/table.php]
- 21ab Cheng M, Ott GR, Anaplastic lymphoma kinase as a therapeutic target in anaplastic large cell lymphoma, non-small cell lung cancer and neuroblastoma, Anticancer Agents Med Chem. 2010 Mar;10(3):236-49 [PUBMED]
- 22 Hydbring P, Larsson LG, Tipping the balance: Cdk2 enables Myc to suppress senescence, Cancer Res. 2010 Sep 1;70(17):6687-91. Epub 2010 Aug 16. [PUBMED]
- 23ab Ordóñez JL, Osuna D, Herrero D, de Alava E, Madoz-Gúrpide J, Advances in Ewing's sarcoma research: where are we now and what lies ahead? Cancer Res. 2009 Sep 15;69(18):7140-50. Epub 2009 Sep 8 [PUBMED]
- 24ab Watowich SS, Liu YJ, Mechanisms regulating dendritic cell specification and development, Immunol Rev. 2010 Nov;238(1):76-92 [PUBMED]
- 25 Doi T, Puri P, Bannigan J, Thompson J, Disruption of GLI3-ZIC3 interaction in the cadmium-induced omphalocele chick model, Pediatr Surg Int. 2010 Nov 11 [PUBMED]
- 26 Introgen [http://www.introgen.com/infotp.html]
- 27 "Atlas of Genetics and Cytogenetics in Oncology and Haematology." (2002) [http://www.infobiogen.fr/services/chromcancer/Genes/P53ID88.html]
- 28ab Vogelstein B, Kinzler KW. "Achilles' heel of cancer?" Nature (2001). 412(6850): 865-866. [PUBMED]
- 29 Soussi T. "The p53 tumor suppresor gene: from molecular biology to clinical investigation." Annals of the New York Academy of Sciences (June 2000). 910: 121-139. [PUBMED]
- 30 Fighting Cancer with Biotechnology. [http://www.biotechinstitute.org]
- 31 Lane DP, et al. "Regulation of p53 stability. The role pf Mdm2 and nuclear export." CRC Laboratories. University of Dundee. (2002). [http://bst.portlandpress.com/bst/027/027668b06.pdf]
- 32 Li-Fraumeni Syndrome. LFS - Genetic Information. OMIM, National Center for Biotechnology Information. Accessed 10/2/2010 [http://www.ncbi.nlm.nih.gov/omim/151623]
- 33 Senturk S, Yao Z, Camiolo M, Stiles B, Rathod T, Walsh AM, Nemajerova A, Lazzara MJ, Altorki NK, Krainer A, Moll UM, Lowe SW, Cartegni L, Sordella R. p53¿ is a transcriptionally inactive p53 isoform able to reprogram cells toward a metastatic-like state. Proc Natl Acad Sci U S A. 2014 Aug 12;111(32):E3287-96. Epub 2014 Jul 29. [PUBMED]
- 34 Igenetics. Russell PJ. Published by Benjamin Cummings: San Francisco, CA: 2002.
- 35 Polyak K, Xia Y, Zweier J, Zinzler K, Vogelstein B. "A model of p53-induced apoptosis." Nature (1997). 389: 300-305. [PUBMED]
- 36 Dameron KM, Volpert OV, Tainsky MA, Bouck N. "Control of angiogenesis in fibroblasts by p53 regulation of angiogenesis of thrombospondin-1." Science (1994). 265: 1582-1584. [PUBMED]
- 37 Zambetti M. "Mdm-2: 'big brother' of p53." J Cell Biochem (1997). 64(3): 343-352. [PUBMED]
- 38 Geradts J, Ingram CD. "Abnormal expression of cell cycle regulatory proteins in ductal and lobular carcinomas of the breast." Modern Pathology (2000). 13(9): 945-953. [PUBMED]
- 39 Knudson, Alfred G. "Mutation and Cancer: Statistical Study of Retinoblastoma." <i>Proceedings of the National Academy of Sciences of the United States of America.</i> 68 (1971): 820-823. [PUBMED]
- 40 Poznic M. Retinoblastoma protein: a central processing unit. J Biosci. 2009 Jun;34(2):305-12. [PUBMED]
- 41 Herwig S, Struss M. "The retinoblastoma protein: a master regulator of cell cycle, differentiation, and apoptosis." Eur J Biochem (1997). 246: 581-601. [PUBMED]
- 42 Singh P, Chan SW, Hong W. "Retinoblastoma protein is functionally distinct from its homologues in affecting glucocorticoid receptor-mediated transcription and apoptosis." Journal of Biological Chemistry (2001). 276: 13762-13770. [PUBMED]
- 43 Witkiewicz AK, Ertel A, McFalls JM, Valsecchi ME, Schwartz G, Knudsen ES. RB-pathway Disruption is Associated with Improved Response to Neoadjuvant Chemotherapy in Breast Cancer. Clin Cancer Res. 2012 Jul 20. [Epub ahead of print] [PUBMED]
- 44 Zini N, Trimarchi C, Claudio P, Stiegler P, Marinelli F, Maltarello M, La Sala D, De Falco G, Russo G, Ammirati G, Maraldi N, Giordano A, Cinti C. "pRb2/p130 and p107 control cell growth by multiple strategies and in association with different compartments within the nucleus." Journal of Cellular Physiology (2001). 189: 34-44. [PUBMED]
- 45 DePhino R. "Transcriptional repression: the cancer-chromatin connection." Nature (1998). 391(6667): 533-536. [PUBMED]
- 46 Magnaghi-Jaulin L, Groisman R, Naguibneva I, Lorrain S, LeVillain JP, Troalen F, Trouche D, Harel-Bellan A. "Retinoblastoma protein represses transcription by recruiting a histone deacetylase." Nature (1998). 391(6667): 601-605. [PUBMED]
- 47 Iwamoto M, Ahnen DJ, Maltzman F, Maltzman T. "Expression of beta-catenin and Full Length APC Protein in Normal and Neoplastic Colonic Tissues." Carcinogenesis (2000). 21(11): 1935-1940. [PUBMED]
- 48 Fultz KE, Gerner EW. "APC-Dependent Regulation of Ornithine Decarboxylase in Human Colon Tumor Cells." Molecular Carcinogenesis (2002). 34:10-18. [PUBMED]
- 49 Morin PJ, Vogelstein B, Kinzler KW. "Apoptosis and APC in Colorectal Tumorigenesis." Proc. Natl. Acad. Sci. USA (1996). 93: 7950-7954. [PUBMED]
- 50abcde Welcsh PL, King MC. "BRCA-1 and BRCA-2 and the Genetics of Breast and Ovarian Cancer." Human Molecular Genetics (2001). 10(7): 705-713. [http://hmg.oupjournals.org/cgi/content/full/10/7/705] [PUBMED]
- 51ab Narod SA, Boyd J. "Current Understanding of Epidemiology and Clinical Implications of BRCA-1 and BRCA-2 Mutations for Ovarian Cancer." Current Opinion in Obstetrics and Gynecology (2002). 14: 19-26. [PUBMED]
- 52abcd Aoki K, Taketo MM. Adenomatous polyposis coli (APC): a multi-functional tumor suppressor gene. J Cell Sci. 2007 Oct 1;120(Pt 19):3327-35. [http://www.ncbi.nlm.nih.gov/pubmed/17881494] [PUBMED]
- 53abc Johan H. van Es,1 Rachel H. Giles, and Hans C. Clevers. The Many Faces of the Tumor Suppressor Gene APC. Experimental Cell Research 264, 126¿134 [http://www.ncbi.nlm.nih.gov/pubmed/11237529] [PUBMED]
- 54 Greenberg RA. Recognition of DNA double strand breaks by the BRCA1 tumor suppressor network. Chromosoma. 2008 Aug;117(4):305-17. Epub 2008 Mar 28. [http://www.ncbi.nlm.nih.gov/pubmed] [PUBMED]
- 55 Hashemi J, Lindstrom MS, Asker C, Platz A, Hansson J, Wiman KG. "A melanoma-predisposing germline CDKN2A mutation with functional significance for both p16 and p14ARF." Cancer Lett (2002). 180(2): 211-21. [PUBMED]
- 56 Pierceall WE, Reale MA, Candia AF, et al. Expression of a homologue of the deleted in colorectal cancer (DCC) gene in the nervous system of developing Xenopus embryos. Dev Biol 1994; 166:654¿665. [http://www.ncbi.nlm.nih.gov/pubmed] [PUBMED]
- 57 Keino-Masu K, Masu M, Hinck L, et al. Deleted in colorectal cancer (DCC) encodes a netrin receptor. Cell 1996;87:175¿185. [http://www.ncbi.nlm.nih.gov/pubmed] [PUBMED]
- 58 Grady WM. Making the case for DCC and UNC5C as tumor-suppressor genes in the colon. Gastroenterology. 2007 Dec;133(6):2045-9. [http://www.ncbi.nlm.nih.gov/pubmed/18054576] [PUBMED]
- 59 Gallo A, Cuozzo C, Esposito I, Maggiolini M, Bonofiglio D, Vivacqua A, Garramone M, Weiss C, Bohmann D, Musti AM. "Menin uncouples Elk-1, JunD and c-Jun phosphorylation from MAP kinase activation." Oncogene (2002). 21(42): 6434-45. [PUBMED]
- 60 Yachida S, Iacobuzio-Donahue CA. The pathology and genetics of metastatic pancreatic cancer. Arch Pathol Lab Med. 2009 Mar;133(3):413-22. [http://www.ncbi.nlm.nih.gov/pubmed/19260747] [PUBMED]
- 61 de Caestecker MP, Piek E, Roberts AB. "Role of transforming growth factor-beta signaling in cancer." J Natl Cancer Inst (2000). 92(17): 1388-402. [PUBMED]
- 62 Heldin CH, Landström M, Moustakas A. Mechanism of TGF-beta signaling to growth arrest, apoptosis, and epithelial-mesenchymal transition. Curr Opin Cell Biol. 2009 Apr;21(2):166-76. Epub 2009 Feb 23. [http://www.ncbi.nlm.nih.gov/pubmed/19237272] [PUBMED]
- 63 Starker LF, Carling T. Molecular genetics of gastroenteropancreatic neuroendocrine tumors. Curr Opin Oncol. 2009 Jan;21(1):29-33. [http://www.ncbi.nlm.nih.gov/pubmed/19125015] [PUBMED]
- 64 Rocco JW, Sidransky D. p16(MTS-1/CDKN2/INK4a) in cancer progression. Exp Cell Res. 2001 Mar 10;264(1):42-55. [http://www.ncbi.nlm.nih.gov/pubmed] [PUBMED]
- 65 Johannessen CM, Reczek EE, James MF, Brems H, Legius E, Cichowski K. The NF1 tumor suppressor critically regulates TSC2 and mTOR. Proc Natl Acad Sci U S A. 2005 Jun 14;102(24):8573-8. [http://www.ncbi.nlm.nih.gov/pubmed] [PUBMED]
- 66 Gladden AB, Hebert AM, Schneeberger EE, McClatchey AI. The NF2 Tumor Suppressor, Merlin, Regulates Epidermal Development through the Establishment of a Junctional Polarity Complex. Dev Cell. 2010 Nov 16;19(5):727-39. [http://www.ncbi.nlm.nih.gov/pubmed] [PUBMED]
- 67 Giono LE, Manfredi JJ. The p53 tumor suppressor participates in multiple cell cycle checkpoints. J Cell Physiol. 2006 Oct;209(1):13-20. [http://www.ncbi.nlm.nih.gov/pubmed/16741928] [PUBMED]
- 68 Backman S, Stambolic V, Mak T. "PTEN function in mammalian cell size regulation." Curr Opin Neurobiol (2002). 12(5): 516. [PUBMED]
- 69 Yamasaki L. Role of the RB tumor suppressor in cancer. Cancer Treat Res. 2003;115:209-39. [http://www.ncbi.nlm.nih.gov/pubmed/12613199] [PUBMED]
- 70 Kaelin WG Jr. "Molecular basis of the VHL hereditary cancer syndrome." Nat Rev Cancer (2002). 2(9):673-82. [PUBMED]
- 71 Bernstein C, Bernstein H, Payne CM, Garewal H. "DNA repair/pro-apoptotic dual-role proteins in five major DNA repair pathways: fail-safe protection against carcinogenesis." Mutat Res (2002). 511(2): 145-78. [PUBMED]
- 72 Lee RC, Feinbaum RL, Ambros V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell. 1993 Dec 3;75(5):843-54. [PUBMED]
- 73 Bartel DP. MicroRNAs: target recognition and regulatory functions. Hum Mol Genet. 2005 Apr 15;14 Spec No 1:R121-32 [PUBMED]
- 74 Kusenda B, Mraz M, Mayer J, Pospisilova S. MicroRNA biogenesis, functionality and cancer relevance. Biomed Pap Med Fac Univ Palacky Olomouc Czech Repub. 2006 Nov;150(2):205-15. [PUBMED]
- 75ab Zhang B, Pan X, Cobb GP, Anderson TA. microRNAs as oncogenes and tumor suppressors. Dev Biol. 2007 Feb 1;302(1):1-12. Epub 2006 Aug 16. [PUBMED]
- 76ab Boyd SD. Everything you wanted to know about small RNA but were afraid to ask. Lab Invest. 2008 Jun;88(6):569-78. Epub 2008 Apr 21. [PUBMED]
- 77 Nikitina EG, Urazova LN, Stegny VN. MicroRNAs and human cancer. Exp Oncol. 2012;34(1):2-8. [PUBMED]
- 78abc Schoof CR, Botelho EL, Izzotti A, Vasques Ldos R. MicroRNAs in cancer treatment and prognosis. Am J Cancer Res. 2012;2(4):414-33. Epub 2012 Jun 28. [PUBMED]
- 79abc Corsini LR, Bronte G, Terrasi M, Amodeo V, Fanale D, Fiorentino E, Cicero G, Bazan V, Russo A. The role of microRNAs in cancer: diagnostic and prognostic biomarkers and targets of therapies. Expert Opin Ther Targets. 2012 Apr;16 Suppl 2:S103-9. Epub 2012 Mar 23. [PUBMED]
- 80abc Zen K, Zhang CY. Circulating microRNAs: a novel class of biomarkers to diagnose and monitor human cancers. Med Res Rev. 2012 Mar;32(2):326-48. doi: 10.1002/med.20215. Epub 2010 Nov 9. [PUBMED]
- 81ab Liu X, Liu L, Xu Q, Wu P, Zuo X, Ji A. MicroRNA as a novel drug target for cancer therapy. Expert Opin Biol Ther. 2012 May;12(5):573-80. Epub 2012 Mar 20. [PUBMED]
- 82 Neilson JR, Sharp PA. Small RNA regulators of gene expression. Cell. 2008 Sep 19;134(6):899-902. [PUBMED]
- 83 Dang CV. Links between metabolism and cancer. Genes Dev. 2012 May 1;26(9):877-90. [PUBMED]
- 84 Bayley JP, Devilee P. The Warburg effect in 2012. Curr Opin Oncol. 2012 Jan;24(1):62-7. [PUBMED]
- 85 Gao P, Sun L, He X, Cao Y, Zhang H. MicroRNAs and the Warburg Effect: New Players in an Old Arena. Curr Gene Ther. 2012 Aug 1;12(4):285-91. [PUBMED]
- 86 Yuxia M, Zhennan T, Wei Z. Circulating miR-125b is a novel biomarker for screening non-small-cell lung cancer and predicts poor prognosis. J Cancer Res Clin Oncol. 2012 Jul 18. [Epub ahead of print] [PUBMED]
- 87 Wang H, Tan G, Dong L, Cheng L, Li K, Wang Z, Luo H. Circulating MiR-125b as a marker predicting chemoresistance in breast cancer. PLoS One. 2012;7(4):e34210. Epub 2012 Apr 16. [PUBMED]
- 88 Cho WC. MicroRNAs as therapeutic targets and their potential applications in cancer therapy. Expert Opin Ther Targets. 2012 Aug;16(8):747-59. Epub 2012 Jun 13. [PUBMED]
- 89 Cittelly DM, Finlay-Schultz J, Howe EN, Spoelstra NS, Axlund SD, Hendricks P, Jacobsen BM, Sartorius CA, Richer JK. Progestin suppression of miR-29 potentiates dedifferentiation of breast cancer cells via KLF4. Oncogene. 2012 Jul 2. doi: 10.1038/onc.2012.275. [Epub ahead of print] [PUBMED]
- 90 Schreiber R, Mezencev R, Matyunina LV, McDonald JF. "Evidence for the role of microRNA 374b in acquired cisplatin resistance in pancreatic cancer cells." Cancer Gene Ther. 2016 May 27. [PUBMED]