癌症基因
细胞分裂是由一系列严密控制的流程来完成的。这些流程取决于一些基因的正常转录(transcription)与翻译(translation)。如果这些流程出现异常,则可导致细胞生长的失控。现在认为,人类基因组大约有30,000个基因,其中少部分基因似乎在肿瘤的预防、发生与进展方面起着十分重要的作用。在许多不同类型的恶性肿瘤中,这些基因出现功能异常或功能丧失。
对于已鉴别出的基因,可根据它们在细胞中的正常功能,分为如下两大类:
- 第一类基因是它们的蛋白质产物能刺激或提高细胞的分裂能力与生存能力。这一类基因还包括那些能抑制细胞死亡从而在肿瘤生长上起了一定作用的基因。
- 第二类基因是它们的蛋白质产物能直接或间接阻止细胞分裂或导致细胞死亡。
第一类基因的正常“版本”叫做原癌基因(proto-oncogenes)。然而,这类基因的“突变” 版本或“受损”版本则成了癌基因(oncogenes)。
第二类基因可叫做肿瘤抑制基因(tumor suppressors)。
请注意正文中与“基因”有关的名词是否是“斜体”字型。按照惯例,若基因名词为“斜体”,则表示“基因”本身;若不是“斜体”,则表示“该基因所产生的蛋白质”。例如,p53表示“基因”;而p53则表示“该基因所产生的蛋白质”。
本页的话题有:
癌基因(Oncogenes)
肿瘤抑制基因与癌基因对人体的作用可以用汽车来做比喻。原癌基因就如同下面动画中的汽车油门,控制着汽车的运动。当汽车处于正常状态时,只有踩下油门时汽车才能开动。在正常细胞中,细胞内、外的信号都能控制癌基因的活性。在下面动画中,这些信号用"X"型的生长因子和踩油门的脚来表示。
有缺陷的癌基因就像是油门始终处于“开”的状态,此时不再需要某种信号来激活这些基因,这辆汽车无论是踩或不踩油门都会向前行驶。
把这个比喻用于细胞:即使没有信号指示细胞分裂,细胞仍会持续地分裂下去。我们的每个基因都有两套,对于癌基因来说,只要其中一套基因有缺陷就会导致细胞的持续分裂。
目前已经有大量的基因被鉴定为原癌基因,其中有许多基因负责提供导致细胞分裂的阳性信号。一些原癌基因作用于调节细胞死亡。正如在本节介绍所述,癌基因(也就是这些基因的缺陷版本)可以导致细胞无管制的分裂。即使在没有正常促生长信号,如生长因子的情况下,这种增长仍可以发生。癌基因活动的一个主要特点是一套缺陷版本就可以导致细胞生长失控。这与肿瘤抑制基因完全不同,肿瘤抑制基因必须需要两套基因都有缺陷才会导致细胞异常分裂。
迄今,那些被确定的原癌基因在细胞中有许多不同的功能。尽管他们的正常功能有差异,这些基因的突变体形式(致癌版本)均导致无管制的细胞分裂。突变体蛋白通常情况下能保留一些自己原有的功能,但对正常形态下的该做出反应的管理信号不再敏感了。在下文中,我们选取了一些与许多癌症相关的癌基因,并加以详细地描述。
HER-2/Neu
HER-2/neu (即erbB-2)是一种能为人类表皮生长因子受体2型编码的基因。在某些正常细胞上有中等含量的这种受体。顾名思义,这种受体参与了细胞对生长因子的反应。如下图所示,在适当条件下这种基因与生长因子结合则可刺激细胞分裂。
在人类乳腺癌病例中,高达30%的病例具有HER-2/neu 基因的扩增。HER-2/neu基因复制本数目的增加可导致细胞表面HER-2蛋白的表达增高,从而导致细胞增生速度加快(如下图所示)。1目前认为,基因扩增会影响肿瘤生长与扩散的能力,也会影响肿瘤对治疗的反应。该基因的过度表达可使肿瘤更具侵袭性,但也可使肿瘤对一些化疗药物更加敏感。 2 关于基因扩增的更多信息。
HER-2/neu与癌症治疗
HER-2/neu基因的过度表达对化疗药物有效性的影响尚未清楚。目前已开展了几项研究,其目的是确定HER2蛋白对化疗药物有效性到底有什么样的影响。最近的一项研究是让140例原发性乳腺肿瘤患者使用不同浓度的两种化疗药物组合。 研究结果显示,HER-2/neu表达较强的细胞比表达较弱的细胞更易受到化疗药物的影响,肿瘤生长受抑的程度更为明显。HER-2/neu扩增似乎让肿瘤细胞对化疗药物获得敏感性,而不是让它们形成耐药性。化疗药物攻击的是正在复制基因的细胞,HER-2/neu扩增导致复制速度加快。因此,可以得出这样一个结论:由于细胞分裂速度加快,HER2过度表达的肿瘤细胞将被更有效的杀死。然而,由于出现了一些与之矛盾的结果(即对化疗药物不敏感),HER-2/neu扩增的真正作用尚未完全清楚。2
还有一些研究显示,HER-2/neu的过度表达与雌激素受体阴性、肿瘤细胞分化低、病人存活率下降有关。3 显然,这种原癌基因在几种恶性肿瘤的发生上起了重要作用,但是这方面的研究还远远没有结束。
抗体治疗与HER2
癌症治疗一直是针对那些HER2蛋白过度表达的癌症细胞。Genentech公司研制的曲妥单抗 (Herceptin®)是一种人源化单克隆抗体抗体,其能与HER2蛋白结合,阻断其活性,从而阻止细胞的过度增生。下面的动画显示了这个过程。最近,Herceptin®和化疗药物的配合使用去治疗HER-2/neu基因扩增的恶性肿瘤。3, 1关于癌症的抗体治疗的更多信息。
RAS
RAS基因的产物参与了控制激酶的信号传导路径,以及能控制基因转录,从而调节细胞的生长与分化。为了打“开”这条路径,ras蛋白必须与细胞内的三磷酸鸟苷(GTP)分子结合。为了“关”闭这条路径,RAS蛋白必须裂解GTP分子。RAS基因的改变可以使RAS蛋白发生变化,从而使RAS蛋白不再具有裂解GTP分子和释放GTP分子的能力。这样的改变导致这条信号传导路径始终处于“开”通的状态。4这种“开”的信号导致细胞的生长和增生。因此ras的过度表达与扩增导致持续的细胞增殖,这是肿瘤发生的关键一步。5细胞分裂由“正”与“负”信号的平衡来调控。当ras转录增加时,过多的基因蛋白积蓄在细胞内;此时,细胞分裂的“正”信号则强于“负”信号。
RAS基因的点突变通常导致RAS从原癌基因向癌基因的转化。这样的功能改变对细胞的影响是多方面的,因为RAS参与了许多控制细胞分裂与死亡的信号传导路径。目前研制的抗肿瘤药物正是针对这样的RAS依赖性路径。然而,此类药物要应用于临床还得进行许多研究才能实现。6
突变的RAS基因在如下器官的恶性肿瘤中已得到鉴定: 胰腺(90%)、结肠(50%)、肺(30%)、甲状腺(50%)、膀胱(6%)、卵巢(15%)、乳腺、皮肤、肝脏、肾脏、某些类型的白血病。4 在将来,有可能使用ras来鉴定某些恶性肿瘤。胰腺肿瘤一直难于诊断,但是在随粪便排出的胰腺细胞的DNA 里,将RAS基因突变鉴定出来,则可帮助临床医师鉴别胰腺炎与胰腺癌。4
MYC
MYC蛋白是一种转录因子,它控制着几种基因的表达。在许多不同的恶性肿瘤中都发现有MYC基因的突变,如伯基特淋巴瘤,B细胞白血病和肺癌。MYC癌基因家族可通过基因重排和基因扩增被激活。基因重排涉及到染色体的断裂与重排。这过程可波及到大量DNA,影响到多个基因。基因或基因群的移动可发生在一个染色体内,也可发生在不同染色体之间。这样的移动常常导致基因表 的改变和细胞功能的改变。
染色体易位是一种基因重排,在8号与14号染色体之间的易位常导致MYC基因的过度表达,最终导致B细胞淋巴瘤。下面的动画显示了两个不同染色体之间发生易位的情形。
MYC蛋白在细胞中的含量是十分重要的,因为MYC蛋白活性的平衡要靠另一种蛋白与MYC蛋白抗衡。因此,这两种蛋白中的任何一种的含量增加都会破坏这种平衡,从而影响细胞分裂。7
在上面的视频中,杰拉德·埃文博士讨论了MYC蛋白在癌症中的作用。观看完整的埃文博士采访。(Watch the full interview with Dr. Evan.)
MYC活性的增加有时会导致细胞的程序化死亡,但这种保护机制似乎会因另一种癌基因bcl-2的存在而遭到破坏,bcl-2可阻止myc所诱导的细胞凋亡(apoptosis)。7
SRC
SRC是最先发现的癌基因。SRC是在作为劳斯肉瘤病毒(RSV)的转化试剂(致癌试剂)时被鉴定出来的。这种病毒可感染鸡和其它动物。RSV是一种逆转录病毒(retrovirus)。它能够感染细胞,并能将自己的基因“插入”到这个细胞的DNA中,很快导致癌症的发生。因此,这种病毒被称作急性转化病毒。当鸡被感染后,常在两周内出现大块肿瘤。研究人员发现,来自于RSV中的某个基因的蛋白质可导致细胞以异常方式生长。在人类基因组也发现了相对应的原癌基因。这种人类基因,如果被激活为癌基因后,也会以相似的方式致癌。
SRC蛋白是一种酪氨酸激酶。激酶是一种可将磷酸基转移到靶分子上的酶。这个过程非常重要,因为去除或增添磷酸基可使生物分子发生改变,从而调节细胞的活动。磷酸基的增添或去除就像开关一样,调控着靶分子的活性。src蛋白可改变几种靶分子,从而传递信号给细胞核,有助于细胞活动的调节。7
目前已发现至少9种SRC基因。由于这些基因所产生的mRNA进过不同的处理加工,所以生成了至少14种不同的蛋白质。在大多数细胞中会发现低含量的C-SRC。但是在某些类型的恶性肿瘤中,C-SRC会被过度表达,比如人类神经母细胞瘤、小细胞性肺癌、结肠癌、乳腺癌、横纹肌肉瘤。4
端粒酶
由于DNA复制过程的特性,染色体的末端(称作“端粒”)会在每次细胞分裂过程中变短。变短的染色体限制了细胞可进行分裂的次数。当端粒缩短到一定程度时,如果细胞继续进行DNA复制,就会丢失重要的遗传物质。此时,正常细胞会进入细胞衰老过程,或出现生长骤停。在这两种情况下,细胞不再继续分裂。
然而,癌细胞却有能力继续复制它们的DNA,不进入衰老期。在许多癌症中,这种无限制的细胞分裂能力是由于一种叫端粒酶的产生所致。这种酶能维护染色体的末端,从而防止它变短。端粒酶是一种存在于胎儿发育时期的正常蛋白质。然而在成人的大多数细胞中,这种酶已不再存在,因为这种酶的基因不再被表达(被转录和翻译)。但是在某些癌细胞中,为这种酶编码的基因被重新激活,最终导致复制失控。
以下动画显示了两种染色体,右侧是具有活性的端粒酶,左侧是不具活性的端粒酶。
在癌细胞中,即使不存在端粒末端转移酶活性,染色体也不会缩短,这种现象可由其它机制解释。因为只有保持端粒的长度,才能使细胞分裂不受限制。hTERT是为端粒酶的活性成分编码的基因,被认为是原癌基因,因为它的异常表达可导致细胞生长失控。
BCL-2
BCL-2 (即B细胞淋巴瘤基因2)蛋白与细胞膜以及细胞膜的活性密切相关。BCLl-2蛋白是调控细胞凋亡的复杂信号系统的一个组成部分。细胞凋亡可由多种信号诱导,包括DNA的不可修复性损伤。这种细胞以自杀的方式阻止了细胞损伤的进一步扩展。Bcl-2可阻止细胞凋亡。8因此,其过度表达可阻止损伤细胞的凋亡,这样就导致了突变细胞系的持续分裂,最终导致癌症的发生。此外,BCL-2的过度表达也可导致一些癌症的转移。9
如果细胞凋亡的控制受到破坏,那么诱导细胞凋亡的抗癌药物就不会像以前那样有效了。 所以一些药物正在被开发,作用于抑制BCL-2基因的表达,让其它抗癌药物更有效(而且剂量更少)。其中一种药物是Genasense,其为一种反义核苷酸。这种药物在一期实验中可降低BCL-2蛋白的产生,目前正在进行二期和三期试验,可作为多种恶性肿瘤治疗的辅助性药物。10关于反义药物(antisense drugs)的更多信息。
此外,还有一些药物是间接地减少BCL-2蛋白含量,比如全反式维甲酸、紫杉醇、长春新碱、多西紫杉醇。这几种药物常与其它化疗药物联合使用。还有一些新的药物 (尚未进行人体实验),比如“BCL-2结合多肽”能导致BCL-2蛋白失活、抗霉素A能与“Bcl-2相关蛋白”结合。11
既然BCL-2基因的过度表达会影响癌症治疗的成功率,所以确定其功能是否正常则成为一种有价值的诊断依据。这种原癌基因可通过易位导致基因过度表达,继而激活为癌基因。在许多不同的癌症中发现了高含量的BCL-2蛋白。12
EGFR
表皮生长因子受体(EGFR)是一种跨膜蛋白。它有一部分在细胞外,然后穿过细胞表面,伸出一部分到细胞质。它的功能是作为一种蜂窝天线。当特定的蛋白质与EGFR结合时,两个表皮生长因子受体蛋白会彼此粘连。EGFR的例子有:表皮生长因子或转化生长因子α。结合的两个蛋白质整体被称为二聚体,而二聚体的形成会激活受体,从而导致自身磷酸化过程。这两个蛋白质会互相添加一种小分子化学物质,叫做磷酸基团。 EGFR的激活可以提高细胞增殖和存活的活性。13
在许多癌症中,EGFR基因发生突变或过度活跃,这些癌症有乳腺癌,肺癌,食道癌,头颈部癌。14在癌细胞和在肿瘤中存在的其它细胞中,EGFR的过度活动会导致血管生长,过度细胞分裂,增强细胞存活和细胞运动,从而导致癌症的扩散。 13
目前针对EGFR突变使用的靶向疗法有:单克隆抗体和酪氨酸激酶抑制剂(TKI)。一些抗体,例如西妥昔单抗(Erbitux®)和帕尼单抗(Vectibix®),如果和EGFR的细胞外部分结合可以防止配体激活受体。而一些TKI,例如吉非替尼(Iressa®)和厄洛替尼(Tarceva®),如果和EGFR的细胞内部分结合可以防止激活过程。13
癌基因一览表
| 癌基因 | 功能 / 激活 | 癌症* | 参考文献 |
| ABL | 通过酪氨酸激酶活性促进细胞生长 | 慢性粒细胞白血病 | 15, 16 |
| AF4/HRX |
融合影响HRX转录因子/甲基转移酶。 HRX也称为MLL,ALL1和HTRX1 |
急性白血病 | 17 |
| AKT-2 | 为丝氨酸/苏氨酸蛋白激酶编码 | 卵巢癌 | 17, 18 |
| ALK |
为酪氨酸激酶受体编码 |
淋巴瘤 |
19 |
| ALK/NPM | 易位产生融合蛋白与核磷蛋白(npm) |
大细胞淋巴瘤 |
19 |
| AML1 | 为一种转录因子编码 |
急性骨髓性白血病 |
17 |
| AML1/MTG8 | 通过易位产生的新融合蛋白 |
急性白血病 |
17 |
| AXL |
为酪氨酸激酶受体编码 |
造血系统恶性疾病 |
17 |
| BCL-2, 3, 6 | 阻断细胞凋亡 (程序化细胞死亡) |
B- 细胞淋巴瘤、白血病 |
16, 17 |
| BCR/ABL | 通过bcr和abl的融合产生的新蛋白质引发不受调节的细胞生长 |
慢性髓细胞白血病、急性淋巴细胞白血病 |
15, 16 |
| c-MYC | 促进细胞增殖和DNA合成的转录因子 |
白血病、乳腺癌、胃癌、肺癌、宫颈癌、结肠癌、神经母细胞瘤、胶质母细胞瘤 |
20 |
| DBL |
鸟嘌呤核苷酸交换因子 |
弥漫性B-细胞淋巴瘤 | 17 |
| DEK/CAN |
融合产生新蛋白 |
急性骨髓性白血病 |
17 |
| E2A/PBX1 |
融合产生新蛋白 |
急性体B细胞白血病 | 17 |
| EGFR | 通过酪氨酸激酶活性触发细胞生长的细胞表面受体 |
鳞状细胞癌 |
15, 16 |
| ENL/HRX | 通过易位t(11; 19)产生的融合蛋白 |
急性白血病 |
17 |
| ERG/TLS | 融合蛋白由t(16:21)易位产生,erg蛋白是转录因子 |
髓细胞白血病 |
16, 17 |
| ERBB |
通过酪氨酸激酶活性触发细胞生长的细胞表面受体 |
胶质母细胞瘤、鳞状细胞癌 |
15 |
| ERBB-2 | 通过酪氨酸激酶活性触发细胞生长的细胞表面受体; 也称为HER2或neu |
乳腺癌、唾液腺癌、卵巢癌 |
15, 18 |
| ETS-1 | 转录因子 |
淋巴瘤 |
21 |
| EWS/FLI-1 | 通过易位t(11:22)产生的融合蛋白 | Ewing肉瘤 | 16, 21 |
| FMS | 酪氨酸激酶 |
肉瘤 |
22 |
| FOS | API的转录因子 |
骨肉瘤 |
16, 5 |
| FPS | 酪氨酸激酶 |
肉瘤 |
22 |
| GLI | 转录因子 |
胶质母细胞瘤 |
23 |
| GSP | 膜相关的G蛋白 |
甲状腺癌 |
5 |
| HER2/neu | 由于基因扩增导致传导激酶的过度表达 |
乳腺癌、宫颈癌 |
15, 5 |
| HOX11 | 转录因子 |
急性 T- 细胞白血病 |
17 |
| HST | 编码成纤维细胞生长因子 |
乳腺癌、鳞状细胞癌 |
17 |
| IL-3 | 细胞信号分子 |
急性前体B- 细胞白血病 |
17 |
| INT-2 | 编码成纤维细胞生长因子 |
乳腺癌、鳞状细胞癌 |
17 |
| JUN | API的转录因子 |
肉瘤 |
18, 5 |
| KIT | 酪氨酸激酶 |
肉瘤 |
18, 5 |
| KS3 | 疱疹病毒编码的生长因子 | Kaposi氏肉瘤 | 5 |
| K-SAM | 成纤维细胞生长因子受体 |
胃癌 |
17 |
| LBC | 鸟嘌呤核苷酸交换因子 |
髓细胞白血病 |
5, 24 |
| LCK | 酪氨酸激酶 |
T- 细胞淋巴瘤 |
5 |
| LMO1, LMO2 | 转录因子 |
T- 细胞淋巴瘤 |
5 |
| L-MYC | 转录因子 |
肺癌 |
17, 18 |
| LYL-1 | 转录因子 |
急性 T- 细胞白血病 |
17 |
| LYT-10 | 转录因子。 也称为NFκB2 |
B- 细胞淋巴瘤 |
5 |
| LYT-10/Cα1 | 通过(10; 14)(q24; q32)lyt-10移位紧邻Cα1免疫球蛋白基因座产生的融合蛋白 | 17 | |
| MAS | 血管紧张素受体 | 乳腺癌 | 5 |
| MDM-2 | 为一种能抑制并降解p53的蛋白质编码 |
肉瘤 |
17, 18 |
| MLL | 转录因子/甲基转移酶(也称为hrx和ALL1) |
急性骨髓性白血病 |
16, 5 |
| MOS | 丝氨酸/苏氨酸激酶 |
肺癌 |
17, 25 |
| MTG8/AML1 | 转录抑制因子与转录因子的融合。 AML1也称为RUNX1。 |
急性白血病 |
17 |
| MYB | 转录因子 |
结肠癌、白血病 |
17 |
| MYH11/CBFB | 通过转录因子融合产生新蛋白质(通过染色体16上的倒位) | 急性骨髓性白血病 | 17 |
| NEU | 酪氨酸激酶。 也称为erbB-2或HER2 | 胶质母细胞瘤和鳞状细胞癌 | 15, 17 |
| N-MYC | 细胞增殖和DNA合成 |
神经母细胞瘤、 视网膜母细胞瘤、肺癌 |
15, 17 |
| OST | 鸟嘌呤核苷酸交换因子 | 骨肉瘤 | 5 |
| PAX-5 | 转录因子 | 淋巴浆细胞样B-细胞淋巴瘤 | 5 |
| PBX1/E2A | 通过t(1:19)易位形成融合蛋白。 转录因子 |
急性前体B-细胞白血病 |
17 |
| PIM-1 | 丝氨酸/苏氨酸激酶 |
T- 细胞淋巴瘤 |
5 |
| PRAD-1 | 为细胞周期蛋白D1编码,参与细胞周期调节。 |
乳腺癌、鳞状细胞癌 |
17 |
| RAF | 丝氨酸/苏氨酸激酶 | 多种癌症类型 | 17 |
| RAR/PML | 通过t(15:17)易位形成融合蛋白。 视黄酸受体。 | 急性早幼粒细胞白血病 | 15, 17 |
| RAS-H | G蛋白。 信号转导。 |
膀胱癌 |
17 |
| RAS-K | G蛋白。 信号转导。 |
肺癌、卵巢癌、膀胱癌 |
17, 18 |
| RAS-N | G蛋白。 信号转导。 |
乳腺癌 |
17 |
| REL/NRG | 2号染色体缺失形成的融合蛋白。转录因子 |
B- 细胞淋巴瘤 |
17, 18 |
| RET | 细胞表面受体。酪氨酸激酶 |
甲状腺癌、多发性内分泌瘤2型 |
15, 17 |
| RHOM1, RHOM2 | 转录因子 |
急性 T- 细胞白血病 |
17 |
| ROS | 酪氨酸激酶 |
肉瘤 |
5 |
| SKI | 转录因子 | 癌 | 5 |
| SIS | 生长因子 |
神经胶质瘤、纤维肉瘤 |
5 |
| SET/CAN | 通过9号染色体重排形成的融合蛋白。蛋白定位 | 急性骨髓性白血病 | 5, 26 |
| SRC | 酪氨酸激酶 |
肉瘤 |
17, 27 |
| TAL1, TAL2 | 转录因子。TAL1也称为SCL |
急性 T- 细胞白血病 |
17, 28 |
| TAN-1 |
通过t(7:9)易位形成Notch细胞受体的改变形式 |
急性 T- 细胞白血病 |
17, 29 |
| TIAM1 | 鸟嘌呤核苷酸交换因子 |
T- 细胞淋巴瘤 |
5, 30 |
| TSC2 | GTP酶激活剂 |
肾癌、脑瘤 |
5, 31 |
| TRK | 受体酪氨酸激酶 |
结肠癌、甲状腺癌 |
17, 32 |
*以上表格列出的癌症类型是与每个癌基因相关的主要癌症类型,但这不是详尽的列表。
了解更多相关信息,请访问癌症基因组解剖项目Cancer Genome Anatomy Project。
本节小结:癌基因
癌基因
- 癌基因是正常细胞基因(原癌基因)的突变形式。
- 原癌基因的蛋白产物可以刺激细胞分裂和/或抑制细胞死亡。
- 原癌基因可以比喻为一辆汽车的油门踏板。
- 通常情况下,内部和外部信号严格控制原癌基因的活性,但是癌基因有缺陷,所以当他们没有收到适当的信号时,也会处于“开”的模式。
- 癌基因也帮助细胞忽视那些防止健康细胞分裂的负信号。
- 致癌基因可导致细胞持续分裂,即使没有接收到任何促生长信号。
- 以下列举了一些已知致癌基因的不同的细胞作用:
- HER-2/neu
- HER-2/neu 给一种细胞表面受体编码,能刺激细胞分裂。
- HER-2/neu基因在人类乳腺癌中可以给扩增高达30%。
- RAS
- Ras的基因产物参与激酶信号传导途径,最终控制基因的转录,调节细胞生长和分化。
- RAS的过度表达和扩增可导致持续的细胞增殖。
- MYC
- Myc蛋白是一种转录因子,它能控制一些基因的表达。
- Myc被证实会参与避免细胞死亡机制。
- MYC致癌基因可能会被基因重排或扩增激活。
- SRC
- SRC是迄今第一个被发现的基因。
- Src蛋白是一种能调节细胞活性的酪氨酸激酶。
- hTERT
- hTERT 编码一种能维持染色体末端的酶,叫做端粒酶。
- 在大多数正常细胞中,端粒酶只有在胎儿发育过程中才存在。
- 在成年细胞中,hTERT的激活会给他们无限分裂的能力。
- hTERT 编码一种能维持染色体末端的酶,叫做端粒酶。
- BCL-2
- Bcl-2蛋白作用于防止细胞死亡(细胞凋亡)。
- BCL-2的过度表达会导致突变细胞的持续分裂。
- HER-2/neu
了解流程:癌基因
“了解流程”是一个测试您知识的教育游戏。开始游戏:
- 从右边栏选择适当的选项,并将它们拖入左边相对应的方框中。请注意,您将只使用六个选项中其中五个来完成游戏。
- 完成后,请点击“查询”来查看您的正确率。
- 对于不正确的答案,可单击“说明”来复习相关信息。
- 如果想要再试一次,请选择“重置”并重新开始游戏。
Please visit us on a larger screen to play this game.
下图列出了上述一些癌基因的功能。其他许多癌基因都具有这些相似的活动。

肿瘤抑制因子
肿瘤抑制基因作用于许多关键的细胞活动过程,如转录、DNA修复、细胞与细胞之间的信息交流。这些基因的功能丧失将会导致细胞行为方式的异常。
继续之前的那个比喻,肿瘤抑制基因就像是汽车的刹车系统。如果你认为肿瘤抑制基因的每个副本都对细胞的“刹车”起作用,那么这个比喻具有一定的说服力。当两套肿瘤抑制基因的副本都起作用时(在下图中用突出显示的基因和停止标志表示),细胞就会停止分裂(像汽车就会停止行驶一样)。
当肿瘤抑制基因的一套副本出现缺陷,细胞还有另一套副本在起作用。就像汽车停车只使用了前刹或后刹,而不是同时使用了前刹和后刹。尽管停车效果没有那么好,但仍然能停下来。所以,当细胞有一套发生缺陷的肿瘤抑制基因副本时,细胞仍能控制它的细胞分裂。但如果另一套肿瘤抑制基因副本也不起作用时,细胞就会失去对细胞分裂的控制。
在下面章节中,我们将介绍视网膜母细胞瘤(retinoblastoma, Rb)的肿瘤抑制基因,这将是一个阐明这个机制的好例子。
所有癌症都显示出一个或多个肿瘤抑制基因和癌基因的改变。在正常细胞中,这两者生成的蛋白质共同控制着细胞分裂,而在癌细胞中,这样的控制则出现异常。
由于这些基因在癌症发生上是十分重要的,在以下章节中将详细介绍一些特异性肿瘤抑制基因、以及与之有关的癌症。参与这些过程的基因几乎每天都在增加,但这里只能介绍部分至今了解得比较多的基因,从中可以理解癌症是如何导致一下细胞功能的失调。
p53简介
p53 (或TP53)基因在1979年被发现,是迄今为止与癌症相关性最强的基因之一。该基因位于17号染色体,其蛋白产物具有转录因子的功能。p53蛋白所控制的基因参与细胞分裂与生存能力。与其它肿瘤抑制基因一样,p53蛋白作用于阻止细胞生长的失控。
p53蛋白与DNA有直接的相互作用,与其它支配细胞行为方式的蛋白也有相互作用。当发现DNA受损或其它细胞受损时,p53立即启动细胞死亡(即细胞凋亡)机制。p53蛋白在维持细胞正常调控方面起着关键性作用,尤其体现在大约半数的肿瘤中,不论其类型或起源如何,都会发现存在着p53基因的缺陷。7, 33在一个人的一生中,都可能出现使p53基因活性消失的突变(散发性突变)或是通过遗传获得这类型突变。
p53基因的发现
在1979 年,科学家们发现了一种新的蛋白质。这种蛋白质能与猿猴病毒40( SV40)中的一种转化蛋白-大型T抗原结合。与正常细胞相比,这种蛋白质在病毒转化的细胞(具致永生和致癌性)里更为常见。该蛋白质和其相应基因被命名为p53,这是根据该蛋白质的质量是53千道尔顿来命名的。 p53基因位于17 号染色体p13位置上。34
虽然p53是继Rb之后发现的第二个肿瘤抑制因子,但是科学家们直到p53发现后10年才了解它在细胞里的作用。7因为p53在被转化的细胞里含量增高,所以科研人员最初认为它的作用类似癌基因。35最初的研究结果也支持这种看法。科学家们发现,当p53基因进入细胞,细胞则发生转化。但是科研人员后来发现,进入到细胞里的p53基因实际上是这种基因的突变型,因为p53基因的正常功能是阻止细胞转化!35, 7
多种证据表明p53基因是肿瘤抑制因子。自1989年以来,p53研究取得了不断进展。 p53蛋白参与了多个细胞过程。然而将p53的临床使用,如鉴定癌细胞的可行性仍有质疑。36目前研究的重心是测试并评估p53临床治疗的可行性,确定它是否能修复p53基因或取代受损的p53基因。37
p53蛋白的功能
p53蛋白在细胞内起着整合的作用。正常情况下,该蛋白在所有类型的细胞内都存在。这种蛋白位于细胞核内,具有转录因子的功能。p53蛋白位于庞大蛋白网络的中心, “监视”着细胞和细胞DNA的健康情况。p53蛋白像一个组织严谨、配合协调的“管弦乐队的指挥”,能及时发现任何一个细胞损伤并加以控制。当监测到损伤时, p53蛋白的功能就是帮助决定:是否修复损伤细胞,或者诱导损伤细胞死亡(即细胞凋亡)。38
作为转录因子,p53促进一组靶基因的转录。在这组基因中,p21是最重要的一种基因。p21基因的产物是细胞因子依赖性激酶的一种负性调控剂。这种酶在细胞周期的正常运转与最终的细胞分裂中具有关键性作用。7 通过促进p21基因的转录,p53蛋白可阻止细胞增生。这样的阻止作用可以给细胞一个修复的机会。如果出现严重DNA损伤,p53蛋白可协助启动细胞死亡过程。严重DNA受损细胞的死亡对机体是有利的,因为这样阻止了有害突变细胞的持续性复制。
如本节简介里所述,所有癌细胞都含有肿瘤抑制基因与癌基因的联合突变。从细胞里将功能性p53(即基因组的“卫兵”) 去除,则可导致更多DNA损伤的蓄积,更多DNA受损伤的细胞的持续分裂。
p53基因突变是癌细胞中最常见的基因改变。这种突变除了在个体的生长与发育过程中出现(散发性突变),还有一些突变出现在某些类型的恶性肿瘤中, 这些肿瘤与受损p53复制本的遗传有关。比如Li-Fraumeni家族性肿瘤综合征就常与多种各样的恶性肿瘤有关。 39此外,有几种病毒已经进化而具备了使p53蛋白失活的途径。
由于这种蛋白在调节细胞分裂中起到核心作用,大量的科研致力于开发恢复p53基因功能的一种安全的方法。
在2014年,研究人员发现了p53基因的另一种转录剪接形式,其能推动癌症发展和扩散。新的形式被称为p53Ψ。这项工作是在细胞培养物、小鼠抗体、兔抗体中进行的。40
p53蛋白的异常与癌症发展
一个缺乏功能性p53的细胞可能发生或者不发生癌变。相应地,一个拥有正常p53功能的细胞可能最终导致癌细胞的形成。正如在上节基因突变中所讨论的,一个细胞的DNA必须发生一些改变才可能发生突变。p53的其中一个功能是监视细胞DNA的状态。p53可以和一系列其他蛋白质合作,来识别和修复受损的DNA。受损DNA所激发的细胞反应有:修复,停止细胞分裂和细胞死亡。p53基因的损坏确实会增加癌症发展的可能性。请记住,由于p53基因是一种肿瘤抑制基因,只有该基因的两份拷贝都失活才会有作用。以下列举了几种p53失活的方法:
基因突变
p53基因的改变对基因的活性有不同的作用,而这取决于基因被改变的位置。
- 突变可能发生在调控区域。基因的这些区域控制着基因转录的频率和时间,这个区域被称为启动。在启动子区域发生的突变可以导致细胞减少或丢失p53。41
- 突变可能发生在基因的蛋白质编码区,这种突变有以下几种方式来影响基因的表达(或者蛋白质的活性):
- 转录因子p53的活性会降低。这将会影响到p53靶基因的表达。p53的靶基因包括p21(一种参与细胞周期调节的蛋白质),Bax(一种诱导细胞凋亡的蛋白质),血小板反应蛋白1(一种血管生成的抑制剂)。42, 43
- p53蛋白的改变会使它变得更易降解。如果细胞中的p53蛋白被被降解的速度高于正常速度,那么它们将不再具有正常的肿瘤抑制功能。44
病毒失活
p53的其中一个功能是“守卫"基因组。病毒感染会引入外源DNA进入细胞,而p53会与其它蛋白质一起,负责对细胞内的外源DNA做出反应,如停止细胞分裂和激发细胞死亡。但是,几种病毒已经进化出试p53蛋白失活的方法来避免这些反应。这样的例子有猿猴病毒40(SV40)。细胞被感染了SV40后会有病毒蛋白在细胞质内产生。其中一个生成的蛋白质叫做大T抗原。这种蛋白质的功能是与p53蛋白结合,并使它失活。其他病毒,如肝炎和人乳头瘤病毒也会产生类似的蛋白质。
如果细胞丢失了正常工作的p53,细胞将会持续分裂,即使有DNA损伤。在没有p53的情况下,基因的不稳定性,体现在基因突变和非整倍性的可能性会增加。基因损伤的增加会导致缺陷的肿瘤抑制基因和癌基因的积累。45
视网膜母细胞瘤基因Rb的简介
视网膜母细胞瘤(retinoblastoma,Rb)基因可为一种改变转录因子活性的蛋白质编码。通过与转录因子的相互作用, Rb蛋白可间接调控基因表达。除了这种功能,Rb蛋白和与它有密切关系的一些蛋白还有几种其它的功能,虽然有关证据不是很多。 Rb蛋白和这些与它有密切关系的蛋白参与了细胞分裂过程的调控。46
Rb基因的突变发生在多种恶性肿瘤中。在这方面研究得最多的肿瘤之一是视网膜母细胞瘤, Rb基因就是根据这种眼部肿瘤的名字来命名的。该病常见于幼儿。已鉴定有如下两种不同类型:
- 散发型: 可影响任何人,是患者一生中所出现的基因改变(突变)所导致的疾病。
- 家族型: 患者从父母一方通过遗传而获得缺陷基因复制本。患者的每个细胞中的基因复制本有一个正常,有一个出现缺陷。47
与其它肿瘤抑制基因一样,该肿瘤的基因表型不太明显,除非基因的两个复制本都受到损伤。虽然不可能在任何一个细胞中都会出现Rb基因的突变,但是在我们机体(包括眼睛)的许多细胞内可能会出现该基因的继发性突变。家族型患者可能合并有其它不同形式的肿瘤,尤其是骨肉瘤。与Rb基因突变有关的其它肿瘤有:肺癌、乳腺癌、膀胱癌。48
Rb基因与癌症的详细介绍
Rb基因最初是在家族性遗传形式的视网膜母细胞瘤中发现的。这种癌症主要影响眼睛,并且在儿童中最常见。在这种基因的家族型中,患者的细胞中已经具有一个突变的Rb基因,所以只需要另一个Rb基因的突变就能使细胞缺乏功能性的Rb蛋白。虽然突变并不常见,而且一个特定基因发生突变的机率是相当小的,但是我们的身体中有大量的细胞,这使得这种小概率的基因突变事件至少在几个细胞中发生。如果这些细胞能够以不受控制的方式生长,则会引发癌症。因此家族性视网膜母细胞瘤的病人通常会有多个肿瘤。而在散发型视网膜母细胞瘤的案例中,患者的细胞具有Rb基因的两个功能性基因复制本,因此丧失Rb功能需要在同一细胞中同时发生两个突变。所以散发型视网膜母细胞瘤的患者通常只发展一个肿瘤。此外,家族性视网膜母细胞瘤的病人更可能发生肿瘤复发。
研究人员发现在家族性视网膜母细胞瘤的患者中,他们的骨肉瘤丢失了Rb活性。骨肉瘤意味着如果癌症发生转移,那么近一半的可能是病人具有这种疾病的家族性遗传形式。 此外,女性得乳腺癌的机率也与Rb功能有联系。通常Rb能够调节G1细胞周期检查点,而研究表明一些乳腺癌在该检查点中失调。这意味着Rb在这种情况下是疾病的促成因子。此外,Rb也对其它癌症有促进作用,例如小细胞和非小细胞肺癌。
Rb的功能
Rb基因对维持细胞周期的正常功能有着重要的作用。细胞对它们环境中的各种信号作出不同的反应,这些信号能调控它们的生长、分裂、休眠、 细胞凋亡。如果这些信号遭到破坏,则可导致细胞生长的失控,最终导致癌症的形成。细胞分裂过程的调控涉及到不同信号的整体作用。
Rb基因产物(pRb)在正常情况下,具有抑制生长的作用。它能够与转录因子结合,从而抑制转录因子。因此,Rb蛋白能够间接地控制许多基因的表达。其中的一些基因能产生驱动细胞分裂的蛋白质。所以,Rb蛋白可作用于减慢或阻止细胞的分裂。49 调节蛋白(如Rb蛋白)的改变会对细胞乃至整个机体产生明显的影响。Rb蛋白除了能够调节细胞周期之外,还对细胞凋亡有一定影响。细胞凋亡是一个非常重要的细胞功能,它能够使受损细胞进入程序化死亡过程。如果细胞发生的突变是不可修复的,这个细胞就会通过凋亡被清除。这种机制还有助于清除那些有生长失控潜能,而最终成长为癌细胞的细胞。任何能够使凋亡机制削弱或失去的细胞功能改变,都可能对整个细胞群造成损害。50 几种不同类型的基因损伤都可以使 Rb 基因失去活性。使Rb蛋白功能完全丧失的突变(即无效突变)常见于那些丢失功能性Rb蛋白的细胞。
实际上,Rb的丢失可能在某些情况下帮助患者对化疗做出反应。研究表明,乳腺癌患者如果缺乏Rb功能,会对新辅助化疗做出更好的反应(新辅助化疗也就是他们在手术前接受的化疗)。51
Rb功能的详细介绍
最近的研究表明,Rb除了有调节细胞生长和细胞凋亡的功能之外,与Rb相关的蛋白质具有不同的活动,而这取决于细胞周期的阶段和这些蛋白质在细胞核里的位置。此外,还有些研究表明,像Rb一样的蛋白质能调节核糖体核糖核酸和转录核糖核酸的转录过程。这就是说,pRb能控制细胞内的“转录”过程和“转录后”的过程。52
pRb除了充当转录因子外,它还可能有肿瘤抑制作用。Rb蛋白已被证明与染色质修饰蛋白有关,如组蛋白脱乙酰酶(HDAC)。据认为,组蛋白脱乙酰酶通过从组蛋白那里除去乙酰基而对转录过程造成影响。这种修饰导致DNA与细胞核小体之间更加紧密的关系。53 DNA与组蛋白更强烈的相互作用使得转录因子(如 E2F)与它在DNA内的靶位结合更加困难。虽然Rb在该过程中所起的作用仍然不清楚,但是有研究表明至少部分组蛋白脱乙酰酶在Rb缺乏情况下不会正常发挥功能。54
APC简介
APC (腺瘤性结肠息肉病) 基因的突变与遗传性和散发性结肠癌的发生有着非常密切的关系。正如我们在下一部分要介绍的那样, APC蛋白像很多肿瘤抑制蛋白一样,可控制细胞分裂过程中起关键作用的基因表达。
大多数结肠癌病例被诊断是进展缓慢的疾病,病程可长达数年。 在5号染色体上,APC 基因的失活被认为是导致细胞增生加快和结肠息肉形成的主要原因。在正常结肠细胞转变为潜在的肿瘤“种子”细胞的过程中,会发生几种基因的改变。在许多病例中, APC基因的突变被认为是发生最早的一些改变。这种说法可以通过以下证据来间接的证明,即通过检测因遗传而获得APC突变基因的患者。这些病人患有家族性腺瘤性息肉病,患者的结肠内长满了息肉。每个息肉都有可能发展成为癌症,所以这些具有遗传突变的个体患癌的可能性会高得多。这与在“Rb基因”章节中所介绍的视网膜母细胞瘤的遗传形式十分相似。不同的是,这类患者不需要细胞发生两个体细胞突变来导致 APC失去功能,而是任何细胞的一个基因突变就可导致重大问题。
通过对癌症不同阶段的细胞突变进行比较,可建立一部分结肠癌发生基因突变的可能顺序。在这个顺序模型中,APC基因突变发生在第一步,产生了具有高度增殖性的细胞,而这些细胞形成了息肉,最终发展成为癌症。7
APC的功能
由于功能性APC蛋白的缺乏可导致细胞分裂的加快,那么具有正常功能的APC蛋白则可以某种方式抑制细胞分裂。APC蛋白与β-连环蛋白 ( beta-catenin ) 形成复合物可导致β-连环蛋白的降解( β-连环蛋白是一种转录因子 ) 。如果缺乏APC蛋白,过多的β-连环蛋白就会在细胞核内积聚。β-连环蛋白与细胞核内的另一种蛋白结合可形成一种复合物,这种复合物会和 DNA 结合,从而启动了几种基因的转录。这种复合物中的一个靶基因叫做 c-myc,它是一种已知的癌基因。55C-myc本身就是几种基因的转录因子,它控制着细胞的生长和分裂。因此,APC基因突变可导致一系列的连锁反应,从而最终加速了细胞分裂。 下面显示了 APC 功能的图解。
当然,许多其它因子也可影响基因及其产物的表达,但 APC 基因的突变似乎与β-连环蛋白和 c-myc的多少有关,β-连环蛋白和 c-myc数量的增多可导致细胞的增生速度加快。56
研究显示,对缺乏APC 蛋白的结肠癌细胞添加APC 蛋白质有减退肿瘤细胞成长的功能。而成长的减退是由于细胞凋亡的增加,这就意味着 APC 有控制细胞死亡并和成长的功能。57 所以,APC基因的丢失会对调节细胞成长与细胞死亡的平衡有一定的影响,因此APC基因控制着细胞数量。
肿瘤抑制因子:BRCA
BRCA蛋白具有多种功能。其中一种重要功能是修复DNA损伤。这还参与了基因表达的调节。BRCA-1基因与另一种肿瘤抑制基因:p53的激活有关,其靶基因是p21。
BRCA蛋白也与转录因子和其它转录成分有着相互的作用,以控制其它几种基因的活性。58当BRCA基因失活时,DNA修复和基因调节则会受到影响。DNA损伤的加剧会生成一些聚集了关键基因突变的细胞,因而导致恶性肿瘤细胞的形成。缺乏BRCA基因的细胞常出现染色体断裂、严重的非整倍体 以及中心体扩增。
从分子水平上,BRCA-1基因和BRCA-2基因的结构解释了它们对突变的易感性。它们包含着高比例的重复DNA,这在人类基因中是十分罕见的。这种高度重复的 DNA 可导致基因组的不稳定和重排。58
多项研究已经证实,BRCA基因产物的缺乏与散发性或遗传性恶性肿瘤的发生密切相关。58
BRCA基因与卵巢癌的详细介绍
BRCA基因是根据它与乳腺癌的联系而命名的,而这类基因的突变也与卵巢癌有关。卵巢癌的遗传方式和散发规律与乳腺癌相似,但也有一些差异。遗传性卵巢癌多有血清组织学异常,呈“中、低分化”,具浸润性生长特征,通常在晚期发现。此外,BRCA突变基因携带者有较高的输卵管病变发生率。无论病人是否为突变基因携带者,那些良性或低度恶性潜能的卵巢肿瘤不应视为浸润性卵巢癌的前期。59
BRCA的功能
BRCA-1 和 BRCA-2 的基因突变与部分乳腺癌和卵巢癌有关。这两种基因在细胞内有不同的功能。与之前所介绍过肿瘤抑制基因一样,这两种基因的突变可以是自然发生,也可以是遗传获得。遗传获得 BRCA-1 或 BRCA-2 基因突变的个体更容易患乳腺癌。 BRCA 基因突变的携带者(如果寿命长达 85 岁)在一生中有 80% 的可能性患病乳腺癌。 BRCA-2 突变的携带者在一生中有 10-20% 的可能性患病卵巢癌,而 BRCA-1 突变的携带者则有 40-60% 的可能性患病卵巢癌。这些突变的存在也增加了患前列腺癌、胰腺癌、结肠癌以及其它癌症的可能性。对于任何一个人来说,总患病可能性取决于个体基因与环境危险因素。 5-10% 的乳腺癌病例被认为与 BRCA-1 和 BRCA-2 的基因突变有关。
BRCA基因与雌激素 (Estrogen)
BRCA 基因突变与某些器官组织(如乳腺、卵巢)的癌症有关。这意味着雌激素可能对这些组织的癌症发生上起了一定的作用。雌激素水平的波动(比如在青春期、月经期、妊娠期、绝经期)与癌症的发生相关。在青春期和妊娠期,雌激素水平升高可导致乳腺上皮细胞的增生,这就需要这些细胞对 DNA 修复有更强的能力。如果细胞增殖(即细胞分裂)伴有 DNA 修复效率的下降,则可导致癌症的形成。 在下面的动画中,粉红色的小点表示雌激素。而雌激素可刺激细胞分裂,产生癌细胞。58
如果 BRCA 基因的一个复制本发生突变,这样的突变使仅有的功能性基因不能发挥功能,那么 DNA 修复缺陷则可发生。如果修复基因的两个复制本都失去了功能,那么细胞获得突变的可能性增加,就会导致肿瘤的发生。当一个人遗传获得了有缺陷的 BRCA 基因复制本,那么他的所有细胞都会含有这种突变。此时,如果任何一个细胞内的 BRCA 基因的另一个复制本发生突变,则可导致 DNA 修复困难。如果个体没有因遗传而获得 BRCA 等位基因,则需要有两个单独的突变发生才可导致癌症的出现,并且这两个散发的突变必须出现在同一个细胞内。不过,在同一个细胞内同时发生两个突变的可能性极小,这就解释了为什么癌症通常在人们年龄较大时才会出现。58
关于 BRCA 与雌激素的更多信息,请参考Robert A. Weinberg的 癌症生物学 第3,4,7章节。
BRCA基因与存活率的详细介绍
曾有多种研究去调查确定,BRCA突变基因携带者与散发性肿瘤患者之间在存活率方面有什么差异,但得到的研究结果不大一致。可能是实验设计方案不同或其它什么原因造成,比如对照者(散发性肿瘤患者)与试验者(突变基因携带者)之间匹配不当。然而根据他们所患肿瘤的特征,BRCA突变基因携带者的预后较差。但与散发性肿瘤患者相比,其存活率似乎差不多,可能是肿瘤对化学治疗的反应“比较好”。肿瘤对化学治疗的敏感性增高,部分是因为患者的肿瘤增生速度高。这类肿瘤对某些抗癌治疗比较敏感,如γ射线、顺铂、丝裂霉素 C 。这些治疗能破坏DNA。 受损的DNA在正常情况下由功能性BRCA基因产物修复。如果BRCA基因活性消失,细胞则不能像正常时那样有效修复DNA,细胞最后死亡。BRCA突变基因携带者的非癌性细胞,仍然有功能性BRCA基因,能修复受损 DNA 。59
肿瘤抑制基因一览表
| 肿瘤抑制因子 | 功能 | 癌症 * | 参考文献 |
| APC |
控制特定转录因子的功能。这些转录因子参与一些细胞(包括上皮细胞,淋巴细胞等等)的肿瘤发生,发展和稳态。 APC还涉及细胞增殖和其它细胞活性,例如迁移和粘附。 |
家族性腺瘤性和非遗传性结肠直肠癌 |
60, 61 |
| BRCA1, BRCA2 | DNA损伤修复 | 继承性乳腺癌; 卵巢癌 | 62 |
| CDKN2A | 编码肿瘤抑制基因p16和p14ARF的基因座 | 脑瘤 | 1 63 |
| DCC | Netrin-1受体。 调节细胞增殖和肠上皮细胞凋亡 | 结肠直肠癌 | 64, 65, 66 |
| DPC4 (SMAD4) | 参与发展的转移因子; 影响转移和肿瘤侵袭 | 结肠直肠肿瘤,胰腺肿瘤 | 67, 68 |
| MADR2/JV18 (SMAD2) | 介导生长因子受体的信号。 协助SMAD4转运入细胞核 | 结肠直肠癌 | 69, 70 |
| MEN1 |
为menin蛋白的编码,menin蛋白与转录因子,DNA修复蛋白,细胞骨架蛋白等相互作用,但是其功能未明确定义。 |
多发性内分泌瘤1型 | 71 |
| MTS1 |
细胞周期蛋白依赖性激酶抑制剂; 调节细胞周期从G1期到S期。 |
黑色素瘤 | 72 |
| NF1 | GTP酶激活蛋白(RAS-GAP) | 神经纤维瘤病1型 | 73 |
| NF2 | ERM蛋白; 通过组装蛋白复合物并将它们连接到肌动蛋白来组织质膜。 | 神经纤维瘤病2型 | 74 |
| p53 | 为p21的转录因子编码,p21是一种在G1期阻滞细胞周期的蛋白。 p53作用于整合细胞大小,DNA完整性和染色体复制相关的信号。 | 膀胱癌,乳腺癌,结肠直肠癌,食道癌,肝癌,肺癌,前列腺癌和卵巢癌; 脑肿瘤,肉瘤,淋巴瘤和白血病 | 75 |
| PTEN | 脂质磷酸酶,调节细胞存活 | 考登综合征; 增加乳腺癌和甲状腺癌的风险 | 2 76 |
| Rb | 结合并抑制E2F转录因子。 停止细胞周期进展 | 视网膜母细胞瘤,肉瘤; 膀胱癌,乳腺癌,食管癌,前列腺癌和肺癌 | 77 |
| VHL | 细胞周期调节。 可增加p53的稳定性和活性 | 肾细胞癌 | 1 78 |
| WRN | DNA解旋酶和外切核酸酶。 参与修复DNA断裂。 | 沃纳综合征 | 2 79 |
| WT1 | 转录因子;在发展中起到必不可少的作用 | 维尔姆斯肿瘤(儿科肾癌) | 1 |
* 以上表格列出的癌症类型是与每个肿瘤抑制基因相关的主要癌症类型,但这不是详尽的列表。
了解更多相关信息,请访问癌症基因组解剖项目Cancer Genome Anatomy Project。
1. 《癌基因》,作者:Cooper G,出版社:Jones and Bartlett Publishers,出版日期:1995。
2. 《人类癌症的遗传基础》,作者:Vogelstein B,Kinzler KW,出版社:McGraw-Hill,出版日期:1998。
本节小结:肿瘤抑制基因
肿瘤抑制基因
- 肿瘤抑制基因的蛋白质产物可直接或间接地阻止细胞分裂或导致细胞死亡。
- 肿瘤抑制基因可比喻为汽车的制动系统。
- 肿瘤抑制基因的功能丧失可导致细胞的异常行为。
- 下面介绍了一些关键的肿瘤抑制基因的功能:
- p53
- 一种调节基因对细胞分裂和细胞死亡控制的转录因子。
- 在细胞对DNA损伤的反应中有重要的作用。
- 帮助在修复和诱导细胞死亡之间做出决定。
- Rb
- 作用于改变转录因子活性。
- 通过作为抑制剂来控制细胞分裂。
- APC
- APC蛋白结合并刺激转录因子的降解。
- 功能性APC蛋白的丢失导致细胞分裂的增加。
- BRCA
- BRCA蛋白质具有多种功能,包括修复DNA损伤和基因表达的调节。
- BRCA的异常功能可导致DNA修复和基因调控功能受损。
- p53
MicroRNAs
基因是能够编码RNA的长串DNA。多年来,科学家致力于研究的基因是编码信使RNA(mRNA)的基因,因为RNA用于指导蛋白质的产生。 (参见我们的基因功能部分的概述。)其他RNA,如tRNA,snoRNA,rRNA不用于直接生产蛋白质,但是他们间接的帮助蛋白质的生产。
在1993年,科学家们在一种蠕虫中发现了一种新型的RNA,这种RNA非常短小,并且具有惊人的活性。80 科学实验证明,这种RNA能调节不同基因的活性。 虽然mRNA分子的长度可以有数千个核苷酸,但是这种新型RNA只有几十个核苷酸。 在十年内,许多其他类似于这种小型RNA的实例被陆续发现。 科学实验显示,这些microRNAs (或 miRNAs)能控制多种不同的基因和细胞过程。
另一个惊喜来自于研究人员寻找新的miRNA。 其中有一些来自于它们自己的基因,但是有许多来自于其他基因,通常在非编码区,就是那些不编码蛋白质的区域。 此外,生产出的miRNA不具有功能性。 它们需要几个加工步骤,并最终与蛋白质一起完成它们的基因调节活动。
下图中显示了生产功能性miRNA的两种不同途径(图示来于自Wikimedia Commons)。
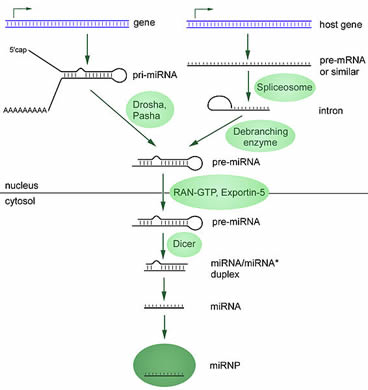
“成熟”过程的最终产物是一种短小的RNA,它能与一组蛋白质(miRNP)相结合。 miRNP用于增加和降低靶基因的活性。 成熟的miRNP可以和靶向的mRNA结合并防止它们用于生产蛋白质。 他们也可以直接造成靶向mRNA的毁坏。 因为在人体细胞中,基因的活性受到严格的控制从而保持平衡,所以miRNA的生产缺陷或者活性缺陷与几种人类疾病(包括癌症)的联系不足为奇。了解更多miRNA在癌症中的作用,请阅读以下页面。81, 82, 83, 84
MicroRNAs和癌症
迄今,microRNA(miRNA)是一种非常常见的RNA,他们能够调节基因对广泛细胞活动的作用。 miRNA的活性的变化可以影响他们靶基因的活性并导致一些可见的变化,如疾病。 癌症是一种改变基因活性的遗传变化,因此miRNA的变化可以影响癌症的发展和/或传播是显而易见的。 事实上,目前miRNA的研究有很大进展,并且已经影响到癌症生物学,检测,诊断和治疗的多种领域。85, 86, 87, 88 以下描述了miRNA是如何影响癌症的。
miRNAs与癌症预防
在食物中发现的化学物质能够影响许多基因的活性,包括那些编码miRNA的基因。 目前,科学家们正积极的探究饮食对miRNA活性的影响从而增加或减少癌症的风险。89
miRNAs作为癌基因和肿瘤抑制基因
因为miRNA控制着基因的活性,所以它们可能是癌基因或肿瘤抑制基因,这取决于它们对细胞生长的影响。 那些能够减慢细胞分裂或引起细胞死亡的miRNA是肿瘤抑制因子,如果丢失这种miRNA,细胞会增加分裂和存活的机率;那些能够增加细胞分裂或存活的miRNA是癌基因。 现在有许多实例显示miRNA能够以这些方式在多种癌症类型中起作用。83, 90, 84, 91
miRNAs作为肿瘤细胞代谢的驱动因子
多年来,科学家们发现癌细胞比正常细胞更多地依赖糖酵解来生产能量,这被称为“Warburg效应”。 这种能量生产途径使癌细胞比其他细胞摄取更多的糖,并且癌细胞还可能会受到体内血糖水平的影响。 此外癌细胞通过糖酵解产生更多的乳酸,这种酸可以改变细胞周围的环境。 而以上这些变化一起可以导致疾病的发展。 最近发现,miRNA可以通过影响肿瘤抑制基因如p53和癌基因如HIF1A的活性来引起Warburg效应。92, 93, 94
miRNA作为癌症检测和诊断的生物标志物
生物标记物是一种间接指示疾病或潜在疾病存在的物质。 例如血液胆固醇水平是一种生物标记物来指示心血管健康。 通常血液检查如PSA测试也用于检查生物标记物。 由于miRNA与癌症相关联,目前研究人员在血液或其他组织中寻找可以作为癌症生物标志物的miRNA。95, 85, 86, 88 他们指出这些miRNA也可以作为耐药性的标记以及用于指导治疗。96
miRNA作为癌症治疗的靶标
作为细胞活性的调节剂,miRNA应该可以作为癌症治疗的靶标。 由于单个miRNA可以控制大量的基因,所以靶向miRNA的药物证明是非常有效的,他们可以立即关闭或打开miRNA的整个通路。97, 85, 86, 88, 90
以乳腺癌为例,许多乳腺癌的生长和存活依赖于女性荷尔蒙雌激素和孕激素。基于这一现象,医生使用抗激素治疗,如他莫昔芬,雷洛昔芬和芳香酶抑制剂。 2012年的一项研究显示,孕激素能导致癌细胞恢复到干细胞一样的状态,使它们更难被治疗。这种细胞行为的变化是由于一组miRNA的抑制而造成的,这组miRNA被称为miRNA 29家族。 研究人员正在寻找方法,使这些miRNA在癌细胞中的活性增强,从而逆转癌细胞中干细胞的特性。98
miRNA作为耐药性的驱动因素
癌症药物有时能够有效地对抗癌症,但这只有在刚开始的时候。 随着时间的推移,患者对药物的敏感性降低。 正是这种耐药性现象使得治疗癌症变得十分困难。 因此,大多数癌症研究正在调查这种耐药性发展的成因和方式。越来越多的证据表明miRNA有助于驱动这种耐药性的变化,许多miRNA的异常水平与耐药性密切相关。 然而当其中一种miRNA恢复到正常水平时,癌症就会恢复对癌症药物的敏感性。99
- 1ab Tsuda H, Akiyama F, Terasaki H, Hasegawa T, Kurosumi M, Shimadzu M, Yamamori S, Sakamoto G. "Detection of Her-2/neu (c-erb B-2) DNA Amplification in Primary Breast Carcinoma." Cancer (2001). 92(12): 2965-2974. [PUBMED]
- 2ab Konecny G, Fritz M, Untch M, Lebeau A, Felber M, Lude S, Beryt M, Hepp H, Slamon D, Pegram M. "Her-2/neu Overexpression and in vitro Chemosensitivity to CMF and FEC in Primary Breast Cancer." Breast Cancer Research and Treatment (2001). 69: 53-63. [PUBMED]
- 3ab Spizzo G, Obrist P, Ensinger C, Theurl I, Dunser M, Ramoni A, Gunsilius E, Eibl G, Mikuz G, Gastl G. "Prognostic Significance of Ep-CAM and Her-2/neu Overexpression in Invasive Breast Cancer." Int. J. Cancer (2002). 98: 883-888. [PUBMED]
- 4abcd Ruddon RW. Cancer Biology. Oxford University Press: New York, 1995.
- 5abcdefghijklmnopqrstuv Vogelstein B, Kinzler KW. The Genetic Basis of Human Cancer. McGraw-Hill: 1998.
- 6 Ahmadian MR. "Prospects for Anti-ras Drugs." British Journal of Haematology (2002). 116(3-I): 511-518. [PUBMED]
- 7abcdefgh Cooper G. Oncogenes. Jones and Bartlett Publishers, 1995. 151-152, 175-176.
- 8 Strasser A, Huang DC, Vaux DL. "The role of the bcl-2/ced-9 gene family in cancer and general implications of defects in cell death control for tumourigenesis and resistance to chemotherapy." Biochim Biophys Acta. 1997 Oct 24;1333(2):F151-78. [PUBMED]
- 9 Fernandez Y, Gu B, Martinez A, Torregrosa A, Sierra A. "Inhibition of Apoptosis in Human Breast Cancer Cells: Role in Tumor Progression to the Metastatic State." Int. J. Cancer (2002). 101: 317-326. [PUBMED]
- 10 Genta Pharmaceuticals [http://www.genta.com]
- 11 Obasaju C, Hudes GR. "Paclitaxel and docetaxel in prostate cancer." Hematol Oncol Clin North Am. 2001 Jun;15(3):525-45. [PUBMED]
- 12 Gross A. "BCL-2 proteins: regulators of the mitochondrial apoptotic program." IUBMB Life. 2001 Sep-Nov;52(3-5):231-6. [PUBMED]
- 13abc Santos, Gilda Da Cunha, Frances A. Shepherd, and Ming Sound Tsao. "EGFR Mutations and Lung Cancer." Annual Review of Pathology: Mechanisms of Disease 6 (2011): 49-69. [http://www.ncbi.nlm.nih.gov/pubmed/20887192] [PUBMED]
- 14 Seshacharyulu, Parthasarathy et al. ¿Targeting the EGFR Signaling Pathway in Cancer Therapy.¿ Expert Opinion on Therapeutic Targets 16.1 (2012): 15¿31. PMC. [http://www.ncbi.nlm.nih.gov/pubmed/22239438] [PUBMED]
- 15abcdefghij Gustave Roussy, Cytogenomics of cancers: From chromosome to sequence, Molecular Oncology, Volume 4, Issue 4, August 2010, Pages 309-322 [PUBMED]
- 16abcdefgh Carlo M. Croce, M.D."Oncogenes and Cancer". N Engl J Med 2008; 358:502-511. [http://www.nejm.org/doi/full/10.1056/NEJMra072367]
- 17abcdefghijklmnopqrstuvwxyz Cooper G. Oncogenes. Jones and Bartlett Publishers, 1995.
- 18abcdefgh Amplified and Over-expressed Genes in Cancer Table. The Institute of Cancer Research. 2009-2010. [http://www.amplicon.icr.ac.uk/table.php]
- 19ab Cheng M, Ott GR, Anaplastic lymphoma kinase as a therapeutic target in anaplastic large cell lymphoma, non-small cell lung cancer and neuroblastoma, Anticancer Agents Med Chem. 2010 Mar;10(3):236-49 [PUBMED]
- 20 Hydbring P, Larsson LG, Tipping the balance: Cdk2 enables Myc to suppress senescence, Cancer Res. 2010 Sep 1;70(17):6687-91. Epub 2010 Aug 16. [PUBMED]
- 21ab Ordóñez JL, Osuna D, Herrero D, de Alava E, Madoz-Gúrpide J, Advances in Ewing's sarcoma research: where are we now and what lies ahead? Cancer Res. 2009 Sep 15;69(18):7140-50. Epub 2009 Sep 8 [PUBMED]
- 22ab Watowich SS, Liu YJ, Mechanisms regulating dendritic cell specification and development, Immunol Rev. 2010 Nov;238(1):76-92 [PUBMED]
- 23 Doi T, Puri P, Bannigan J, Thompson J, Disruption of GLI3-ZIC3 interaction in the cadmium-induced omphalocele chick model, Pediatr Surg Int. 2010 Nov 11 [PUBMED]
- 24 Cerione RA, Zheng Y. "The Dbl family of oncogenes." Curr Opin Cell Biol (1996). 8(2): 216-22. [PUBMED]
- 25 Gorgoulis VG, Zacharatos P, Mariatos G, Liloglou T, Kokotas S, Kastrinakis N, Kotsinas A, Athanasiou A, Foukas P, Zoumpourlis V, Kletsas D, Ikonomopoulos J, Asimacopoulos PJ, Kittas C, Field JK. "Deregulated expression of c-mos in non-small cell lung carcinomas: relationship with p53 status, genomic instability, and tumor kinetics." Cancer Res (2001). 61(2): 538-49. [PUBMED]
- 26 Saito S, Miyaji-Yamaguchi M, Nagata K."Aberrant intracellular localization of SET-CAN fusion protein, associated with a leukemia, disorganizes nuclear export."Int J Cancer. 2004 Sep 10;111(4):501-7. [http://www.ncbi.nlm.nih.gov/pubmed/15239126] [PUBMED]
- 27 Pene-Dumitrescu T, Smithgall TE."Expression of a Src family kinase in chronic myelogenous leukemia cells induces resistance to imatinib in a kinase-dependent manner."J Biol Chem. 2010 Jul 9;285(28):21446-57. Epub 2010 May 7. [http://www.ncbi.nlm.nih.gov/pubmed/20452982/] [PUBMED]
- 28 O'Neil J, Shank J, Cusson N, Murre C, Kelliher M. "TAL1/SCL induces leukemia by inhibiting the transcriptional activity of E47/HEB."Cancer Cell. 2004 Jun;5(6):587-96. [http://www.ncbi.nlm.nih.gov/pubmed/15193261] [PUBMED]
- 29 Pancewicz J, Taylor JM, Datta A, Baydoun HH, Waldmann TA, Hermine O, Nicot C."Notch signaling contributes to proliferation and tumor formation of human T-cell leukemia virus type 1-associated adult T-cell leukemia."Proc Natl Acad Sci U S A. 2010 Sep 21;107(38):16619-24. Epub 2010 Sep 7. [http://www.ncbi.nlm.nih.gov/pubmed/20823234] [PUBMED]
- 30 Masuda M, Maruyama T, Ohta T, Ito A, Hayashi T, Tsukasaki K, Kamihira S, Yamaoka S, Hoshino H, Yoshida T, Watanabe T, Stanbridge EJ, Murakami Y. "CADM1 interacts with Tiam1 and promotes invasive phenotype of human T-cell leukemia virus type I-transformed cells and adult T-cell leukemia cells."J Biol Chem. 2010 May 14;285(20):15511-22. Epub 2010 Mar 9. [http://www.ncbi.nlm.nih.gov/pubmed/20215110] [PUBMED]
- 31 Larson Y, Liu J, Stevens PD, Li X, Li J, Evers BM, Gao T. "Tuberous sclerosis complex 2 (TSC2) regulates cell migration and polarity through activation of CDC42 and RAC1." J Biol Chem. 2010 Aug 6;285(32):24987-98. Epub 2010 Jun 8. [http://www.ncbi.nlm.nih.gov/pubmed/20530489] [PUBMED]
- 32 Liraz Harel, Barbara Costa, Mike Fainzilber On the Death Trk Department of Biological Chemistry, Weizmann Institute of Science, 76100 Rehovot, Israel [http://www.ncbi.nlm.nih.gov/pubmed/20186708] [PUBMED]
- 33 Introgen [http://www.introgen.com/infotp.html]
- 34 "Atlas of Genetics and Cytogenetics in Oncology and Haematology." (2002) [http://www.infobiogen.fr/services/chromcancer/Genes/P53ID88.html]
- 35ab Vogelstein B, Kinzler KW. "Achilles' heel of cancer?" Nature (2001). 412(6850): 865-866. [PUBMED]
- 36 Soussi T. "The p53 tumor suppresor gene: from molecular biology to clinical investigation." Annals of the New York Academy of Sciences (June 2000). 910: 121-139. [PUBMED]
- 37 Fighting Cancer with Biotechnology. [http://www.biotechinstitute.org]
- 38 Lane DP, et al. "Regulation of p53 stability. The role pf Mdm2 and nuclear export." CRC Laboratories. University of Dundee. (2002). [http://bst.portlandpress.com/bst/027/027668b06.pdf]
- 39 Li-Fraumeni Syndrome. LFS - Genetic Information. OMIM, National Center for Biotechnology Information. Accessed 10/2/2010 [http://www.ncbi.nlm.nih.gov/omim/151623]
- 40 Senturk S, Yao Z, Camiolo M, Stiles B, Rathod T, Walsh AM, Nemajerova A, Lazzara MJ, Altorki NK, Krainer A, Moll UM, Lowe SW, Cartegni L, Sordella R. p53¿ is a transcriptionally inactive p53 isoform able to reprogram cells toward a metastatic-like state. Proc Natl Acad Sci U S A. 2014 Aug 12;111(32):E3287-96. Epub 2014 Jul 29. [PUBMED]
- 41 Igenetics. Russell PJ. Published by Benjamin Cummings: San Francisco, CA: 2002.
- 42 Polyak K, Xia Y, Zweier J, Zinzler K, Vogelstein B. "A model of p53-induced apoptosis." Nature (1997). 389: 300-305. [PUBMED]
- 43 Dameron KM, Volpert OV, Tainsky MA, Bouck N. "Control of angiogenesis in fibroblasts by p53 regulation of angiogenesis of thrombospondin-1." Science (1994). 265: 1582-1584. [PUBMED]
- 44 Zambetti M. "Mdm-2: 'big brother' of p53." J Cell Biochem (1997). 64(3): 343-352. [PUBMED]
- 45 Ramel S, Sanchez CA, Schimke K, Neshat K, Cross SM, Raskind WH, Reid BJ. "Inactivation of p53 and the development of tetraploidy in the elastase-SV40 T antigen transgenic mouse pancreas." Pancreas (1995). 11: 213-222. [PUBMED]
- 46 Geradts J, Ingram CD. "Abnormal expression of cell cycle regulatory proteins in ductal and lobular carcinomas of the breast." Modern Pathology (2000). 13(9): 945-953. [PUBMED]
- 47 Knudson, Alfred G. "Mutation and Cancer: Statistical Study of Retinoblastoma." <i>Proceedings of the National Academy of Sciences of the United States of America.</i> 68 (1971): 820-823. [PUBMED]
- 48 Poznic M. Retinoblastoma protein: a central processing unit. J Biosci. 2009 Jun;34(2):305-12. [PUBMED]
- 49 Herwig S, Struss M. "The retinoblastoma protein: a master regulator of cell cycle, differentiation, and apoptosis." Eur J Biochem (1997). 246: 581-601. [PUBMED]
- 50 Singh P, Chan SW, Hong W. "Retinoblastoma protein is functionally distinct from its homologues in affecting glucocorticoid receptor-mediated transcription and apoptosis." Journal of Biological Chemistry (2001). 276: 13762-13770. [PUBMED]
- 51 Witkiewicz AK, Ertel A, McFalls JM, Valsecchi ME, Schwartz G, Knudsen ES. RB-pathway Disruption is Associated with Improved Response to Neoadjuvant Chemotherapy in Breast Cancer. Clin Cancer Res. 2012 Jul 20. [Epub ahead of print] [PUBMED]
- 52 Zini N, Trimarchi C, Claudio P, Stiegler P, Marinelli F, Maltarello M, La Sala D, De Falco G, Russo G, Ammirati G, Maraldi N, Giordano A, Cinti C. "pRb2/p130 and p107 control cell growth by multiple strategies and in association with different compartments within the nucleus." Journal of Cellular Physiology (2001). 189: 34-44. [PUBMED]
- 53 DePhino R. "Transcriptional repression: the cancer-chromatin connection." Nature (1998). 391(6667): 533-536. [PUBMED]
- 54 Magnaghi-Jaulin L, Groisman R, Naguibneva I, Lorrain S, LeVillain JP, Troalen F, Trouche D, Harel-Bellan A. "Retinoblastoma protein represses transcription by recruiting a histone deacetylase." Nature (1998). 391(6667): 601-605. [PUBMED]
- 55 Iwamoto M, Ahnen DJ, Maltzman F, Maltzman T. "Expression of beta-catenin and Full Length APC Protein in Normal and Neoplastic Colonic Tissues." Carcinogenesis (2000). 21(11): 1935-1940. [PUBMED]
- 56 Fultz KE, Gerner EW. "APC-Dependent Regulation of Ornithine Decarboxylase in Human Colon Tumor Cells." Molecular Carcinogenesis (2002). 34:10-18. [PUBMED]
- 57 Morin PJ, Vogelstein B, Kinzler KW. "Apoptosis and APC in Colorectal Tumorigenesis." Proc. Natl. Acad. Sci. USA (1996). 93: 7950-7954. [PUBMED]
- 58abcde Welcsh PL, King MC. "BRCA-1 and BRCA-2 and the Genetics of Breast and Ovarian Cancer." Human Molecular Genetics (2001). 10(7): 705-713. [http://hmg.oupjournals.org/cgi/content/full/10/7/705] [PUBMED]
- 59ab Narod SA, Boyd J. "Current Understanding of Epidemiology and Clinical Implications of BRCA-1 and BRCA-2 Mutations for Ovarian Cancer." Current Opinion in Obstetrics and Gynecology (2002). 14: 19-26. [PUBMED]
- 60 Aoki K, Taketo MM. Adenomatous polyposis coli (APC): a multi-functional tumor suppressor gene. J Cell Sci. 2007 Oct 1;120(Pt 19):3327-35. [http://www.ncbi.nlm.nih.gov/pubmed/17881494] [PUBMED]
- 61 Johan H. van Es,1 Rachel H. Giles, and Hans C. Clevers. The Many Faces of the Tumor Suppressor Gene APC. Experimental Cell Research 264, 126¿134 [http://www.ncbi.nlm.nih.gov/pubmed/11237529] [PUBMED]
- 62 Greenberg RA. Recognition of DNA double strand breaks by the BRCA1 tumor suppressor network. Chromosoma. 2008 Aug;117(4):305-17. Epub 2008 Mar 28. [http://www.ncbi.nlm.nih.gov/pubmed] [PUBMED]
- 63 Hashemi J, Lindstrom MS, Asker C, Platz A, Hansson J, Wiman KG. "A melanoma-predisposing germline CDKN2A mutation with functional significance for both p16 and p14ARF." Cancer Lett (2002). 180(2): 211-21. [PUBMED]
- 64 Pierceall WE, Reale MA, Candia AF, et al. Expression of a homologue of the deleted in colorectal cancer (DCC) gene in the nervous system of developing Xenopus embryos. Dev Biol 1994; 166:654¿665. [http://www.ncbi.nlm.nih.gov/pubmed] [PUBMED]
- 65 Keino-Masu K, Masu M, Hinck L, et al. Deleted in colorectal cancer (DCC) encodes a netrin receptor. Cell 1996;87:175¿185. [http://www.ncbi.nlm.nih.gov/pubmed] [PUBMED]
- 66 Grady WM. Making the case for DCC and UNC5C as tumor-suppressor genes in the colon. Gastroenterology. 2007 Dec;133(6):2045-9. [http://www.ncbi.nlm.nih.gov/pubmed/18054576] [PUBMED]
- 67 Gallo A, Cuozzo C, Esposito I, Maggiolini M, Bonofiglio D, Vivacqua A, Garramone M, Weiss C, Bohmann D, Musti AM. "Menin uncouples Elk-1, JunD and c-Jun phosphorylation from MAP kinase activation." Oncogene (2002). 21(42): 6434-45. [PUBMED]
- 68 Yachida S, Iacobuzio-Donahue CA. The pathology and genetics of metastatic pancreatic cancer. Arch Pathol Lab Med. 2009 Mar;133(3):413-22. [http://www.ncbi.nlm.nih.gov/pubmed/19260747] [PUBMED]
- 69 de Caestecker MP, Piek E, Roberts AB. "Role of transforming growth factor-beta signaling in cancer." J Natl Cancer Inst (2000). 92(17): 1388-402. [PUBMED]
- 70 Heldin CH, Landström M, Moustakas A. Mechanism of TGF-beta signaling to growth arrest, apoptosis, and epithelial-mesenchymal transition. Curr Opin Cell Biol. 2009 Apr;21(2):166-76. Epub 2009 Feb 23. [http://www.ncbi.nlm.nih.gov/pubmed/19237272] [PUBMED]
- 71 Starker LF, Carling T. Molecular genetics of gastroenteropancreatic neuroendocrine tumors. Curr Opin Oncol. 2009 Jan;21(1):29-33. [http://www.ncbi.nlm.nih.gov/pubmed/19125015] [PUBMED]
- 72 Rocco JW, Sidransky D. p16(MTS-1/CDKN2/INK4a) in cancer progression. Exp Cell Res. 2001 Mar 10;264(1):42-55. [http://www.ncbi.nlm.nih.gov/pubmed] [PUBMED]
- 73 Johannessen CM, Reczek EE, James MF, Brems H, Legius E, Cichowski K. The NF1 tumor suppressor critically regulates TSC2 and mTOR. Proc Natl Acad Sci U S A. 2005 Jun 14;102(24):8573-8. [http://www.ncbi.nlm.nih.gov/pubmed] [PUBMED]
- 74 Gladden AB, Hebert AM, Schneeberger EE, McClatchey AI. The NF2 Tumor Suppressor, Merlin, Regulates Epidermal Development through the Establishment of a Junctional Polarity Complex. Dev Cell. 2010 Nov 16;19(5):727-39. [http://www.ncbi.nlm.nih.gov/pubmed] [PUBMED]
- 75 Giono LE, Manfredi JJ. The p53 tumor suppressor participates in multiple cell cycle checkpoints. J Cell Physiol. 2006 Oct;209(1):13-20. [http://www.ncbi.nlm.nih.gov/pubmed/16741928] [PUBMED]
- 76 Backman S, Stambolic V, Mak T. "PTEN function in mammalian cell size regulation." Curr Opin Neurobiol (2002). 12(5): 516. [PUBMED]
- 77 Yamasaki L. Role of the RB tumor suppressor in cancer. Cancer Treat Res. 2003;115:209-39. [http://www.ncbi.nlm.nih.gov/pubmed/12613199] [PUBMED]
- 78 Kaelin WG Jr. "Molecular basis of the VHL hereditary cancer syndrome." Nat Rev Cancer (2002). 2(9):673-82. [PUBMED]
- 79 Bernstein C, Bernstein H, Payne CM, Garewal H. "DNA repair/pro-apoptotic dual-role proteins in five major DNA repair pathways: fail-safe protection against carcinogenesis." Mutat Res (2002). 511(2): 145-78. [PUBMED]
- 80 Lee RC, Feinbaum RL, Ambros V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell. 1993 Dec 3;75(5):843-54. [PUBMED]
- 81 Bartel DP. MicroRNAs: target recognition and regulatory functions. Hum Mol Genet. 2005 Apr 15;14 Spec No 1:R121-32 [PUBMED]
- 82 Kusenda B, Mraz M, Mayer J, Pospisilova S. MicroRNA biogenesis, functionality and cancer relevance. Biomed Pap Med Fac Univ Palacky Olomouc Czech Repub. 2006 Nov;150(2):205-15. [PUBMED]
- 83ab Zhang B, Pan X, Cobb GP, Anderson TA. microRNAs as oncogenes and tumor suppressors. Dev Biol. 2007 Feb 1;302(1):1-12. Epub 2006 Aug 16. [PUBMED]
- 84ab Boyd SD. Everything you wanted to know about small RNA but were afraid to ask. Lab Invest. 2008 Jun;88(6):569-78. Epub 2008 Apr 21. [PUBMED]
- 85abc Schoof CR, Botelho EL, Izzotti A, Vasques Ldos R. MicroRNAs in cancer treatment and prognosis. Am J Cancer Res. 2012;2(4):414-33. Epub 2012 Jun 28. [PUBMED]
- 86abc Corsini LR, Bronte G, Terrasi M, Amodeo V, Fanale D, Fiorentino E, Cicero G, Bazan V, Russo A. The role of microRNAs in cancer: diagnostic and prognostic biomarkers and targets of therapies. Expert Opin Ther Targets. 2012 Apr;16 Suppl 2:S103-9. Epub 2012 Mar 23. [PUBMED]
- 87 Nikitina EG, Urazova LN, Stegny VN. MicroRNAs and human cancer. Exp Oncol. 2012;34(1):2-8. [PUBMED]
- 88abc Zen K, Zhang CY. Circulating microRNAs: a novel class of biomarkers to diagnose and monitor human cancers. Med Res Rev. 2012 Mar;32(2):326-48. doi: 10.1002/med.20215. Epub 2010 Nov 9. [PUBMED]
- 89 Ross SA, Davis CD. MicroRNA, nutrition, and cancer prevention. Adv Nutr. 2011 Nov;2(6):472-85. Epub 2011 Nov 3. [PUBMED]
- 90ab Liu X, Liu L, Xu Q, Wu P, Zuo X, Ji A. MicroRNA as a novel drug target for cancer therapy. Expert Opin Biol Ther. 2012 May;12(5):573-80. Epub 2012 Mar 20. [PUBMED]
- 91 Neilson JR, Sharp PA. Small RNA regulators of gene expression. Cell. 2008 Sep 19;134(6):899-902. [PUBMED]
- 92 Dang CV. Links between metabolism and cancer. Genes Dev. 2012 May 1;26(9):877-90. [PUBMED]
- 93 Bayley JP, Devilee P. The Warburg effect in 2012. Curr Opin Oncol. 2012 Jan;24(1):62-7. [PUBMED]
- 94 Gao P, Sun L, He X, Cao Y, Zhang H. MicroRNAs and the Warburg Effect: New Players in an Old Arena. Curr Gene Ther. 2012 Aug 1;12(4):285-91. [PUBMED]
- 95 Yuxia M, Zhennan T, Wei Z. Circulating miR-125b is a novel biomarker for screening non-small-cell lung cancer and predicts poor prognosis. J Cancer Res Clin Oncol. 2012 Jul 18. [Epub ahead of print] [PUBMED]
- 96 Wang H, Tan G, Dong L, Cheng L, Li K, Wang Z, Luo H. Circulating MiR-125b as a marker predicting chemoresistance in breast cancer. PLoS One. 2012;7(4):e34210. Epub 2012 Apr 16. [PUBMED]
- 97 Cho WC. MicroRNAs as therapeutic targets and their potential applications in cancer therapy. Expert Opin Ther Targets. 2012 Aug;16(8):747-59. Epub 2012 Jun 13. [PUBMED]
- 98 Cittelly DM, Finlay-Schultz J, Howe EN, Spoelstra NS, Axlund SD, Hendricks P, Jacobsen BM, Sartorius CA, Richer JK. Progestin suppression of miR-29 potentiates dedifferentiation of breast cancer cells via KLF4. Oncogene. 2012 Jul 2. doi: 10.1038/onc.2012.275. [Epub ahead of print] [PUBMED]
- 99 Schreiber R, Mezencev R, Matyunina LV, McDonald JF. "Evidence for the role of microRNA 374b in acquired cisplatin resistance in pancreatic cancer cells." Cancer Gene Ther. 2016 May 27. [PUBMED]